Reinigungsvalidierung in der Pharmazeutischen Industrie
Abstract
Die Reinigung von Equipment ist eine Disziplin, die ein hohes Maß an chemischem, toxikologischem, pharmakologischem und verfahrenstechnischem Know-how erfordert. Nur wenn alle fachlichen Disziplinen zusammenarbeiten, lässt sich ein möglichst wirtschaftliches und dennoch zuverlässiges Verfahren für multipurpose-Anlagen etablieren und validieren. In diesem Artikel sollen chemische, toxikologische und pharmakologische Aspekte zur Planung und Auslegung des Reinigungsverfahrens und verfahrenstechnische Bewertung von Bracketing und Probenahme diskutiert werden.
1 Einleitung und Historie
Der Grundstein für die Notwendigkeit der Reinigungsvalidierung wurde mit der Einführung der Penicilline gelegt. Im FDA „Guide to In spections- Validiation of Cleaning Processes“ 7/93 [5] heißt es, dass die meisten Produktrückrufe aufgrund von Kreuzkontamination durch Penicilline hervorgerufen wurden. Im Hinblick auf die Reinigung waren auch die mangelnde Hygiene und fehlende Staub Kontrolle ein weiterer Grund für die Behörden, regulatorische Bestimmungen festzulegen. Der FDA Guide stammt aus dem Jahr 1993 und enthält alle wesentlichen Grundzüge der Reinigungsvalidierung. Der Guide bezieht sich auf die Herstellungsprozesse der chemischen und der biotechnologischen Pharmaindustrie. Die Gründe zur Entstehung dieses Guides liegen in zuvor aufgetretenen Skandalen wie z.B. Pestizidrückständen in Arzneimitteln und mangelhafter Beweisführung für die Abwesenheit von Rückständen in einer multi-purpose Anlage in der auch Steroide hergestellt wurden.
Heute muss für jede Reinigungsvalidierung eine risikobasierte Betrachtung sämtlicher Stoffe, die in das nachfolgende Produkt gelangen können, erfolgen. Hierzu zählen Rückstände von Wirkstoffen, Reinigungsmitteln und möglichen Abbauprodukten. Um mögliche Kreuzkontaminationen im Folgeprodukt zu vermeiden, werden Akzeptanzkriterien definiert. Diese werden gemäß ICH Q9 [9] risikobasiert für jeden einzelnen Wirkstoff einer Mehrzweckanlage festgelegt. Akzeptanzkriterien für mikrobiologische Grenzwerte wer den nicht genannt, jedoch werden die mikrobiologischen Aspekte in den einschlägigen Regelwerken und Empfehlungen wie dem FDA Guide [5], EU-GMP-Leitfaden, Annex 15 [4] und PIC/S [11] erwähnt.
2 Grenzwerte
für Rückstände von Produkten, Reinigungsmitteln und möglichen Abbauprodukten
Mit dem 2015 revidierten Annex 15 EU GMP Leitfaden wurde ein neuer Ansatz für die Betrachtung von möglichen Rückständen veröffentlicht. Mit den Neuerungen sind die bisherigen Akzeptanzkriterien für Produktrück stände, 1/1000 Dosiskriterium und 10-ppm Mengenkriterium nicht mehr allein zu verwenden. Vielmehr sollte das angewandte Kriterium aufgrund einer Risikobewertung der Kontaminanten erfolgen, die neben toxikologischen Betrachtungen auch die Bewertung der pharmakologischen und physikalisch-chemischen Eigenschaften (z.B. Löslichkeiten) beinhalten. Die bisherigen Grenzwerte sollen durch den wissenschaftsbasierten Grenzwert für die tägliche Exposition PDE (Permitted Daily Exposure) oder einen TTC (Threshold of Toxicological Concern)-Wert abgelöst werden. Dieser ist für jeden einzelnen Wirkstoff und jedes Reinigungsmittel zu ermitteln. Für generische Wirkstoffe kann die Ermittlung der Werte über die Beauftragung der Erstellung eines entsprechenden Gutachtens erfolgen.
Stehen die Werte nicht zur Verfügung, z.B. bei Neuentwicklungen, besteht für Arzneimittel-Hersteller die Möglichkeit dass Mengen- oder Dosiskriterium in Verbindung mit dem OEL-Wert (Occupational Exposure Limit) anzuwenden. Hierzu muss der Hersteller nach weisen, dass das bisher verwendete Kriterium größer oder gleich dem jeweiligen OEL-Wert des Wirkstoffes, inklusive Betrachtung des Aufnahmeweges, ist. Der OEL-Wert stellt einen aus toxikologischen Daten hergeleiteten Grenzwert dar.
Gründe dieser Neuerungen sind zum einen, dass das Dosis- und Mengenkriterium willkürlich ohne risikobasierte Ansätze gewählt wurde und keine wissenschaftliche Betrachtungsweise bestand. Der unterschiedliche therapeutische Index von Arzneistoffen fand keine Berücksichtigung. Zudem ist die therapeutische Dosis proportional zum Rückstand, was bei hochdosierten Stoffen zu hohen Produktrück ständen führt. Außerdem müssen Langzeitintoxikationen und teratogene toxische Effekte in die Risikobetrachtung einbezogen werden.
2.1 PDE-Kriterium
PDE(Permitted Daily Exposure) beschreibt die Dosis eines Stoffes, bei der bei täglicher Aufnahme über den gesamten Lebenszeitraum kein negativer Effekt beobachtet wird. Das Akzeptanzkriterium stützt sich auf wissenschaftliche Daten wie klinische oder toxikologische Studien und enthält eine Risikobetrachtung. ADE (Acceptable Daily Exposure) stellt ein Synonym des Kriteriums dar. Die Berechnung der Dosis wird auf Basis von toxikologischen Studien nach der EMA “Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities” durch geführt [3].
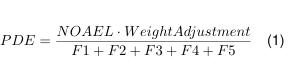
- PDE:Permitted Daily Expose [mg/Tag]
- NOAEL: Höchste Dosis, bei der kein nachteiliger kritischer Effekt zu beobachten ist (No Observed Adverse Effect Level) [mg/(Tag kg)] Weight Adjustment = Standard Körpergewicht 50 kg
- F1:Faktor zur Extrapolation zwischen den Spezies (2-12)
- F2: Faktor zur Unterscheidung zwischen den Spezies (10)
- F3: Faktor zur Berechnung von Kurz zeit/Langzeit Studien
- F4: Faktor bei schwerer Toxizität
- F5:Variabler Faktor wenn kein NOAEL bekannt, sondern PDE von LOEL (Lowest Observed Adverse Effect Level) abgeleitet wird.
Sollte der NOAEL nicht verfügbar sein, kann der LOAEL (Lowest Observed Adverse Effect Level) verwendet werden. Für produktspezifi sche Ausrüstung (Dedicated Equipment) be steht hinsichtlich von Wirkstoffrückständen kei ne generelle Validierungsplicht. Eine Bewertung bezüglich möglichen Abbauprodukten, Reinigungsmittelrückständen und mikrobiologischen Verunreinigungen muss jedoch erfolgen.
Anhand einer Risikobewertung sollte abgewogen werden, ob eine Mehrzweck oder produktspezifischen Anlage zur Herstellung der Arzneimittel verwendet werden kann. Bei der Ermittlung der PDE ist zu berücksichtigen, dass die Route der Administration des Folgeprodukts bekannt sein muss und in die Berechnung des PDEs einbezogen werden müssen. Bei einer subkutanen Anwendung kann z.B. bei vielen Lokalanästhetika eine höhere PDE toleriert werden, als für eine intravenöse Anwendung.
Die Reinigung der Anlagen nach der Benutzung erfordert oft den Zusatz von Reinigungssubstanzen. Auch diese können die Gesundheit der Patienten beeinträchtigen indem sie eine eigene toxische Wirkung entfalten oder die Wirksamkeit des Folgearzneimittels beeinträchtigen. Daher muss sowohl für Wirk- und Hilfsstoffe, als auch für die Reinigungsmittel ein Grenzwert ermittelt werden. Wie auch bei den Wirkstoffen muss dieser Grenzwert erreichbar und messbar sein. Bei einer Reinigungsroutine, die aus mehreren Schritten besteht, sollte man die Analytik auf die Last-to-rinse Substanz beziehen. Auf jeden Fall muss die Zusammensetzung des Reinigungsmittels bekannt sein und der Lieferant eine Langzeitgarantie auf die Rezeptur geben. Als Reinigungsmittel kommen üblicherweise Tenside in Frage, aber auch eine Abfolge von Säuren, Laugen und Komplexierungsmitteln ist üblich.
Wenn die PDE für die geplante Folgeanwendung ermittelt wurde, muss die maximal zulässige Rückstandsmenge MZR (=MACO) für alle Rückstände (Produkte, Abbauprodukte, Reinigungsmittel) berechnet werden.
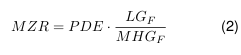
- MZR: Maximal zulässiger (Reinigungsmittel)Rückstand
- LGF: Chargengröße des Folgeproduktes (worst case)
- MHDF: Maximale humantherapeutische (Tages)dosis des Folgeproduktes = Einnahmehäufigkeit x Masse Darreichungsform
Bekannte chemische Strukturen mit unbekannten Toxizitäten können nach Allhenn und Anhalt [1] mit dem TTC-Konzept (Threshold of Toxicological Concern) beurteilt werden. (6) Genotoxische Stoffe oder Stoffe mit sensibilisierendem Potential können nicht über den TTC Wert definiert werden. Hierfür muss der PDE Wert oder die „Limits of Genotoxic Impurities“ [1] herangezogen werden.
2.2 GRAS-Status
Bei Substanzen, die GRAS-Status (generally regarded as safe) gemäß CFR 21 Part 184 [12] haben und keine bekannte PDE vorliegt, kann analog zu ICH Q3C das PDE für Restlösemittel mit niedrigem toxischem Potential PDE = 50 mg zugrunde gelegt werden [10].
2.3 10-ppm-Kriterium
Wenn die Toxizität der Stoffe sehr gering ist oder unbedenkliche Stoffe eingesetzt werden, empfiehlt es sich, dennoch einen „best practise“ Grenzwert festzulegen. Hier empfiehlt es sich das 10 ppm Kriterium als technisch machbaren Grenzwert zu verwenden. Maximal 10 ppm des Vorgängerproduktes dürfen in das Nachfolgeprodukt verschleppt werden. Damit ergibt sich für die Belastung der Folgecharge ein maximaler Wert an Rückstand mmax:
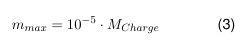
- mmax: Akzeptanzkriterium/ max. zulässiger Rückstand des Vorproduktes [mg]
- MCharge: minimale Chargengröße des Folgeprodukts [kg]
Das 10 ppm Kriterium hat seinen Ursprung in der Lebensmittelindustrie und berücksichtigt nicht die toxikologischen oder pharmakologischen Eigenschaften des Stoffes. Es ist jedoch weiterhin ein nützliches Kriterien, um bei toxikologisch unbedenklichen Stoffen ein Limit zu berechnen.
2.4 Das 1/1000 Dosiskriterium
Auch wenn das toxikologische Kriterium bei der Berechnung der maximalen Rückstandsmenge in Betracht zu ziehen ist, wird in Annex 15 gefordert, die pharmakologischen Eigenschaften und damit auch die Dosierung in die Risikobetrachtung einzubeziehen [4]. Daher sollte man weiterhin die übliche Dosierung des Folgeproduktes ermitteln und in die Diskussion des Grenzwertes einbeziehen.
In der Vergangenheit wurde die übliche bzw. auch die minimale Dosierung mit einem festen Risikofaktor der Grenzwertberechnung zugrunde gelegt. In der Tagesdosis des Folgeproduktes durfte nicht mehr als ein Tausendstel der niedrigsten therapeutischen Tagesdosis des Vorproduktes enthalten sein. Bezogen auf die kleinstmögliche Chargengröße so wie der Anlagenfläche erhält man die maximal zulässige Rückstandsmenge die von der produktberührenden Fläche auf die nächstfolgende Charge übergehen darf:
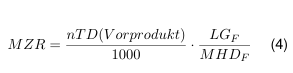
- MZR: Maximal zulässiger (Reinigungsmittel)Rückstand
- nTD: niedrigste therapeutische Dosis des Vorprodukts [mg/d]
- LGF: Chargengröße des Folgeproduktes (worst case)
- MHDF: Maximale humantherapeutische (Tages)dosis des Folgeproduktes = Einnahmehäufigkeit x Masse Darreichungs form
2.5 „Visually-Clean“- Kriterium
Die Anlagenoberfläche muss sichtbar sauber sein. Diese Forderung gilt generell bei der Reinigungsvalidierung. Allerdings ist eine Quantifizierung der Beobachtung „sauber“ schwierig. Der Versuch, das Kriterium für die visuelle Überprüfung zu quantifizieren wurde empirisch mittels „Spiking“-Studien (Verdünnungsstufen) für eine Gruppe von Substanzen ermittelt. Der Grenzwert, bei dem visuell die unter suchten Substanzen lagen, liegt etwa bei bei 4 µg / 100 cm² [6]. Da dieser Wert nur für wenige Produkte getestet wurde, kann er nicht auf alle Produkte übertragen werden. Ein Hersteller muss durch eigene „Spiking“-Studien belegen, ab welcher Konzentration ein Produkt gerade noch visuell erfasst werden kann. Dabei muss die Testoberflächeneigenschaft der Qualität der Anlagenoberfläche aus der Produktion entsprechen. Weiterhin stellen Produkte mit stark färbenden Eigenschaften ein Problem bei der Beurteilung der visuellen Sauberkeit dar. Es wird empfohlen die Unbedenklichkeit von Farbstoffen im Rahmen einer Risikoanalyse zu bewerten und gegeben falls die entsprechen den Produkte mit in die Reinigungsvalidierung einzubeziehen.
Das „Visually-Clean“ Kriterium berücksichtigt weder den Bezug zur Chargengröße noch zur Anlagenfläche. Deshalb ist dieses Kriterium immer im Zusammenhang mit dem PDE Kriterium und ggf. dem 10-ppm Kriterium zu betrachten und kann nicht als alleiniges Kriterium herangezogen werden.
2.6 Worst-case-Konzept
Nach Ermittlung der Grenzwerte wird die Kenntnis der PDE (und ggf. andere Grenz werte) dazu verwendet, produktabhängig einen Grenzwert zu berechnen. Man kann dann für das gesamte Produktspektrum ein worst-case Szenario aus Vorprodukt/Folgeprodukt bestimmen und diesen Grenzwert für die gesamte Reinigungsvalidierung betrachten. Dieser Grenzwert muss verifizierbar sein, d.h. ein validiertes Analysenverfahren muss diesen Grenz wert nachweisen können. Bei Herstellung neu er Produkte auf der Anlage muss die Gültigkeit des worst-case-Szenarios überprüft werden. Bei hochpotenten Wirkstoffen und Arzneimitteln wie Steroide, Antibiotika und Zytostatika ergibt die Risikobetrachtung, dass diese Produkte mit „dedicated equipment“ hergestellt werden müssen, weil z.B. ein ermittelter Grenzwert unterhalb der analytischen Nachweisgrenze liegt oder die Behörde dies bei bestimmten Produktgruppen fordert.
2.7 Umstellung von Dosiskriterium auf PDE
Wurde die Reinigungsvalidierung schon vor 2015 etabliert, mussten seitdem die in der Vergangenheit etablierten Grenzwerte hinterfragt und angepasst werden. Teilweise müssen bessere Reinigungsverfahren etabliert werden, teilweise ist der bisherige Grenzwert aber strenger. Eine Aufweitung von schon etablierten Grenzwerten ist nicht akzeptabel, da man eine technisch machbare Qualität der Reinigung beibehalten muss.
Eine toxikologische Betrachtung des Lokalanästhetikums „Bupivacain“ durch DPhE [8] er gab für die Reinigungsvaldierung, je nach Applikationsweg des Folgeprodukts, unterschiedliche Konsequenzen. Betrachtet wurden zwei Anlagen. Zur Ableitung von Maßnahmen bezüglich der Reinigungsvalidierung wird ein Bewertungsquotient aus dem alten 1/1000-Dosis Kriterium und dem neuen PDE benutzt.
Anlage A Die Folgeprodukte werden ausschließlich subcutan angewendet. Der Bewertungsquotient beträgt 0,2. Es sind keine Maß nahmen erforderlich.
Anlage B Die Folgeprodukte werden meist intravenös angewendet. Der Bewertungsquotient beträgt 3,9. Die Reinigung muss verbessert werden und die Reinigungsvalidierung muss erneut durchgeführt werden.
3 Mikrobiologische Grenzwerte
Während es bei der Betrachtung von Produkt und Reinigungsmittelrückständen auf die Vermeidung von Kreuzkontamination und Verschleppung durch Vorgängerprodukt und Reinigungsprozess ankommt, bezieht sich die Betrachtung der Keimzahl auf die Überwachung und die vorbeugenden Maßnahmen. Solche Maßnahmen sind zum Beispiel, dass das Equipment nach der Reinigung trocken gehalten werden muss. Die Zeiten zwischen Produktionsende und Reinigungsbeginn „dirty-hold time“ haben nicht nur Einfluss auf die Reinigung sondern auch auf die mikrobielle Belastung, was zum Beispiel in der Sterilfertigung eine besondere Rolle spielt. Ebenso müssen die Standzeiten zwischen Reinigungsende und Produktionsbeginn validiert werden. Diese bei den Gegebenheiten haben Einfluss auf das mikrobielle Wachstum. Die Grenzwerte für Keim zahlen an Oberflächen können hierbei aus den Anforderungen aus Annex 1 des EG-GMP Leitfadens [2] bei sterilen Arzneimitteln abgeleitet werden. Eine weitere Orientierung hinsichtlich der mikrobiologischen Reinheit von Arzneimitteln sind die Spezifikationen der jeweiligen Arzneibücher. Für Phytopharmaka gibt das europäische Arzneibuch Hinweise zur mikrobiellen Reinheit von Drogen in den verschiedenen Verarbeitungsstufen. Die mikrobiologichen Grenzwerte für die Reinigungsvalidierung können somit in diesem Fall aus den Produktanforderungen abgeleitet werden.
Eine Sonderstellung stellt hier die Sterilfertigung dar. Obwohl der Sterilisationsnachweis des Equipments durch die Validierung von Sterilisationsprozessen erbracht wird, kann den noch nicht sichergestellt werden, dass Pyrogene bzw. Endotoxine beseitigt wurden. Unter diesem Aspekt ist auch die Mikrobiologie im Rahmen der Reinigungsvalidierung und Standzeitvalidierung von Reinigungsverfahren in der Sterilfertigung nicht außer Acht zu lassen [5, 11].
4 Anlagendesign
Mit Hilfe einer Risikoanalyse soll der Einfluss von produkt-, anlagen- und prozessbezogenen Parametern auf das Reinigungsziel bewertet werden. Um den Aufwand der Reinigungsvalidierung zu reduzieren, kann durch eine Ähnlichkeitsbetrachtung hinsichtlich des Designs der Anlagen und Produkte mit vergleichbaren chemisch-physikalischen Eigenschaften eine Gruppierung, das sogenannte „Bracketing“, vorgenommen werden. Dadurch müssen ähnliche Produkte und Prozesse nicht einzeln validiert werden. Mögliche Kriterien zur Bildung von Gruppen sollen an einem Beispiel eines Rührbehälters dargestellt werden (siehe Tab. 1)
In diesem Beispiel wurde der Fokus auf Konstruktionsmerkmale gelegt, die sich signifikant auf die Reinigung auswirken. Damit konnte eine risikobasierte Rationale für die Zusammenfassung von Anlagen mit unterschiedlichen Konstruktionsmerkmalen dargelegt wer den. Im nächsten Schritt wird dann in jeder Gruppe der worst-case Behälter ermittelt und damit die Reinigungsvalidierung durchgeführt. Für ein erfolgreiches Reinigungsverfahren sind leicht zu reinigende Komponenten unumgänglich. Die reinigungsgerechte Gestaltung der Anlagen und Anlagenteile wird mit dem Hygienic Design beschrieben. Jede produktberührende Oberfläche muss mit dem Reinigungsmittel benetzbar und einfach zu trocknen sein. Bei der hygienegerechten Gestaltung der Anlage tragen auch der Werkstoff und die Oberfläche der Anlage eine entscheidende Rolle zur Reinigbarkeit bei. Beispielsweise können durch eine mangelnde Oberflächenglätte, unsaubere Schweißnähte, ungeeigneten Dichtungskonstruktionen sowie nachträgliche Einbauten massive Probleme bei der Reinigung hervorgerufen werden.
5 Reinigungsverfahren
Neben dem manuellen Reinigungsverfahren besteht auch die Möglichkeit eines automatischen Reinigungsverfahrens (CIP-System). Ein CIP-Reinigungssystem ist allerdings in vielen Fällen nicht realisierbar und zu teuer, weshalb manuell gereinigt werden muss. Dieses ist gegenüber dem CIP-Reinigungssystem hin sichtlich der Reproduzierbarkeit und Validierbarkeit deutlich im Nachteil. Die manuelle Reinigung ist in Ihrer Durchführung stark von der jeweiligen Person abhängig. Deshalb ist nicht nur eine regelmäßige Schulung und die richtige Arbeitsmotivation des Personals, sondern auch die Notwendigkeit einer detaillierten Anweisung, Planung und Überwachung für den Reinigungserfolg entscheidend. Das CIP-System stellt einen kontinuierlichen Prozess dar, der reproduzier- und standardisier bar ist. Der Nachteil liegt darin, dass der Reinigungserfolg aufgrund des geschlossenen Systems schlecht inspizierbar ist. Für die optische Kontrolle des Reinigungserfolgs sowie die Probenahme des Reinigungswassers müssten Bullaugen und Probenahmestellen eingeplant werden. Die Reinigbarkeit von CIP-Systemen kann auch im Rahmen der Qualifizierung mittels einer Bestimmung des Rückstands von Riboflavin erfolgen. Riboflavin weist eine gute Detektierbarkeit auf und ist zudem unbedenklich. Zu bedenken ist auch, dass viele weitere Schritte wie eine Programmierung von Verfahrensparametern, eine Zugriffssicherung oder Archivierung der Daten unternommen werden müssen. CIP-Systeme sind stationäre Anlagen, die fest in die Produktionsanlage integriert sind. Alle auf der Produktionsanlage relevanten Prozesse und Produkte müssen dementsprechend auf das System eingestellt werden.

Die kritischen Mess-, Steuer-, und Regeleinheiten müssen im CIP-System überprüft und regelmäßig rekalibriert werden. Mit Hilfe der in den CIP-Systemen installierten Sprühkugel wird die gesamte Oberfläche der Anlage mit Reinigungslösung benetzt. Die Kugel kann statisch oder drehbar sein. Die Durchflussmenge des Reinigungsmittels ist über den Druck so einzustellen, dass eine Zerstäubung der Lösung vermieden wird. Statisch und dynamisch auftretende Druckverluste können mit einer variabel festgelegten Pumpleistung vermieden werden.
Chemische Eigenschaften des Reinigungs mittels Oft wird mit Wasser gespült, um die Reinigungsmittelanalytik zu umgehen. Das Reinigungsergebnis ist oft schlecht oder sehr viel Wasser wird verbraucht. Hier lohnt sich ein Blick auf die Chemie des zu reinigenden Stoffes. Manchmal kommt eine Komponente (Hilfsstoff) aus der Formulierung in geringer Konzentration als Reinigungszusatz in Frage. Die folgenden Effekte können zum Beispiel für die Reinigung im wässrigen Milieu genutzt werden:
- pH-Verschiebung bei Arzneistoffen, die protonierbar sind
- angehobener Salzgehalt erhöht Löslichkeit schwerlöslicher Stoffe (Aktivität wird verringert)
- Lösungsvermittler/Emulgatoren aus Formulierung von Emulsionen
Der Vorteil dieser Vorgehensweise ist, dass keine zusätzlichen Komponenten zugesetzt werden und damit die Anlage nicht mit zusätzlichen Chemikalien belastet wird, die wieder nachgewiesen werden müssen.
6 Probenahme
In den Plänen zur Reinigungsvalidierung müssen die Orte der Probenahme und eine Begründung für die Auswahl der Orte beschrieben werden. Bei der Probenahme werden zwei an erkannte Arten unterschieden: die direkte und die indirekte Probenahme.
Bei der direkten Probenahme handelt es sich um den Wischtest (Swab), bei dem mittels Wattestäbchen eine definierte Oberfläche mit einem geeigneten Lösungsmittel beprobt wird. Voraussetzung ist hier die Zugänglichkeit der Probenahmestellen worin auch der wesentliche Nachteil liegt: es wird nicht die gesamte Oberfläche beprobt und man kann nicht davon ausgehen, dass die Verunreinigung gleichförmig in einer Anlage verteilt ist. Zudem weist diese Art der Probenahme eine geringe Reproduzierbarkeit auf. Der Vorteil liegt darin, dass man das Ergebnis aus der Analytik direkt einer definierten Stelle in der Anlage zuordnen kann. Auch können schwer lösliche Rückstände mit dem Wischtest erfasst werden.
Die indirekte Probenahme (Rinse) wird bei nicht oder schwer zu erreichenden Stellen, wie Rohrleitungen aber auch Behälterinnenoberflächen und Einbauten, eingesetzt. Der Nach teil der Methode liegt in der Unsicherheit ob alle Rückstände ausgespült worden sind, ob die Rückstände wasserlöslich sind und die Abtragung auch an schlecht zugänglichen Stellen erfolgt. Ein wesentlicher Vorteil liegt je doch in der Möglichkeit eine große Oberfläche zu beproben. Ein weiterer Nachteil der Rinse Methode ist es im Allgemeinen, dass die nach zuweisenden Substanzen stark verdünnt wer den und es dadurch schwer wird, den berechneten Grenzwert analytisch nachzuweisen. Eine weitere Möglichkeit der Probenahme besteht in der Kombination beider Methoden, z.B. eine Beprobung mittels Swab an Stutzen und einer anschließenden Rinse-Beprobung des Gesamtinnenraums. Für beide Probenahmearten ist jeweils die Wiederfindungsrate zu bestimmen.
7 Analytische Methoden
Der Reinigungserfolg einer Methode muss mit analytischen Methoden belegt werden. Im FDA „Guide to inspections validation of cleaning processes“ [5] wird die Notwendigkeit der Spezifität und Sensitivität der analytischen Methoden gefordert. Dabei muss immer beachtet werden, dass Ergebnisse unterhalb der Nachweisgrenze nicht bedeuten, dass in der Probe keine Rückstände vorhanden sind. Das Ergebnis kann nur so gut sein wie die Empfindlichkeit, Spezifität und Genauigkeit des Tests. Daher müssen die analytischen Methoden validiert werden. Folgende Analysenmethoden werden üblicherweise in der Reinigungsvalidierung verwendet.
Spezifische Analysen
- HPLC
- GC
- Farbtests /Fertigkits
- Farbreaktionen auf Proteine (z.B. BCA)
Summenparameter
- TOC
- Leitfähigkeit
- UV/VIS-Spektroskopie
7.1 Analytik für Produkte
HPLC/GC-Analyse: Während der Vorteil der spezifischen Analyse sicherlich in der Spezifität für eine bestimmte Substanz (Wirk- oder Leitsubstanz) liegt, so ist diese Methode in der chemischen Industrie nur für Endprodukte, bzw. nur für den Schritt anwendbar, in dem der Wirkstoff synthetisiert wurde. Daher ist wie bereits der FDA-Guide betont eine spezifische Analyse in der Wirkstoffproduktion nicht praktikabel.
Die spezifischen Methoden haben den Vorteil, dass gezielt spezifische Produkte / Reinigungsmittelkomponenten nachgewiesen werden können. Der Nachteil liegt allerdings darin, dass bei manchen Reinigungsverfahren durch chemische Degradation die tatsächliche vorher vorhandene Menge nicht mehr nachgewiesen werden kann. Hier kann man dann für Analytik der Reinigungsvalidierung eine unspezifischere Bestimmung eines Summenparameters ein setzen.
Falls das Reinigungsverfahren den Wirkstoff zersetzt, wie es z.B. bei der Reinigung von Proteinrückständen mit Natronlauge der Fall ist, reicht eine Risikoanalyse, die den Denaturierungsprozesse bewertet, evtl. kombiniert mit einer Abreicherungstudie.
7.2 Grenzen der Messmethoden
Wenn Säuren oder Laugen als Reinigungsmittel (Zusatz) eingesetzt werden, wird oft der Summenparameter Leitfähigkeit als Meßkriterium für die Abreicherung des Reinigungsmittels verwendet. Daraus ergeben sich folgende Fragen:
- Welche Aussagefähigkeit hat diese Messmethode hier?
- Wenn nach Reinigung mit Purified Water gespült wird und die Leitfähigkeit von Purified Water wieder erreicht ist, sind dann al le Reinigungsmittelrückstände auf ein vertretbares Maß entfernt worden?
Hier muss man im Vorfeld die Grenzen dieses Messverfahrens betrachten und entsprechend auslegen. Das folgende Rechenbeispiel soll die Grenzen des Verfahrens erläutern. Aus den Tabellenwerken [7] ergibt sich eine Nachweisbarkeit von Säuren und Laugen aus den Leitfähigkeiten der Säuren und der maximal zu erwartende Blindwert aus dem Grenzwert für die Leitfähigkeit von Purified Water. Ein Konzentrationsgrenzwert, der kleiner ist, als die Konzentration die sich aus der Annahme ergibt, dass die gesamte Leitfähigkeit am Grenzwert von purified water von dem Reinigungsmittel stammt, gilt als nicht einsetzbar. Als Beispiel soll eine NaOH-Konzentration in WfI berechnet werden. Für Wasser für Injektionszwecke beträgt der Grenzwert der Leitfähigkeit bei 20°C 1,1 µS/cm. Die Leitfähigkeit von NaOH in einer 0,5%igen Lösung beträgt 24,8 µS/cm. Daraus ergibt sich eine Restkonzentration NaOH im WfI von 0,22 mg/l, was ei nem pH-Wert von 8,7 entspricht und damit die Spezifikation von WfI verletzt und daher ungeeignet als Detektionskriterium ist. Das oft verwendete Verfahren „Kessel voll laufen lassen“ würde zu hohe Flüssigkeitsmengen beinhalten und die Aussagekraft der Leitfähigkeitsmessung wäre nicht gegeben. Etwas bessere Nachweisgrenzen erreicht man, wenn man mit Wasser einer geringeren Leitfähigkeit spült und folgende Vorgehensweise einhält. laufen lassen“ würde zu hohe Flüssigkeitsmengen beinhalten und die Aussagekraft der Leitfähigkeitsmessung wäre nicht gegeben. Etwas bessere Nachweisgrenzen erreicht man, wenn man mit Wasser einer geringeren Leitfähigkeit spült und folgende Vorgehensweisen einhält.
- Blindwert Wasser vom aktuellen Spülgang vermessen
- Als Vergleichswert: auf Grenzwert mit Spülwasser verdünnte Reinigungslösung einsetzen.
8 Validierung
Wie in jeder Validierung müssen die Daten aus mehreren Läufen in der Vergangenheit zur Validierung herangezogen werden. Üblicherweise beginnt man mit der Auswertung von drei Läufen und erweitert die Datenlage mit regelmäßigen, geplanten Revalidierungsläufen. Für die Planung der Validierung sind auch Standzeiten einzubeziehen, die sich am geplanten Produktionsablauf orientieren sollten. Seit 2015 [4] ist es zulässig, schon nach dem ersten Validierungslauf mit Verifizierung der Reinigungserfolges die produzierte Charge in den Markt zu bringen. Dies bedeutet insbesondere für Prüfpräparate eine Erleichterung.
9 Fazit
Reinigungsvalidierung ist mehr als „Spülen“, sie erfordert die genaue Betrachtung und Bewertung der einzelnen Schritte und ihrer Leistungsfähigkeit und erfordert ein hohes Maß an interdisziplinärem Wissen
Lorem ipsum dolor sit amet consectetur. Viverra feugiat iaculis aliquam eget sapien sit purus et ultricies. Iaculis nulla egestas risus a. Eget ornare facilisi amet id morbi. Est lectus quisque at vel. In nisi vitae adipiscing augue ante tellus. Enim neque curabitur accumsan tortor senectus. Neque cras fringilla lacinia tristique quis in erat massa.
Literatur
1
ALLHENN, D ; ANHALT, E: Auswahl von Akzeptanzkriterien fr die Reinigungsvalidierung von Mehrzweckanlagen. In: Pharm. Ind. 77 (2015), Nr. 7, S. 1074 1080
2
EUROPEAN COMMISSION: EU Guidelines to Good Manufacturing Practice, Volume 4, Annex 1: Manufacture of Sterile Medicinal Products. 2008
3
EUROPEAN MEDICINES AGENCY: Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities. 2014
4
EUROPEAN COMMISSION: EU Guidelines to Good Manufacturing Practice, Volume 4, Annex 15: Qualification and Validation. 2015
5
FOOD AND DRUG ADMINISTRATION: Guide to Inspections- Validation of Cleaning Processes, 1993
6
FOURMAN, G L. ; MULLEN, M: Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations. In: Pharmaceutical Technology (1993), Nr. 4, S. 54–60
7
HAYNES, William M. (Hrsg.): CRC Handbook of Chemistry and Physics. 91. Baca Raton, FL, 2010
8
HEBENBROCK,K ;HRACH, J: Interner Bericht DPhE: Toxikologisches Assessment von Bupivacain- Ermittlung der PDE. 2015
9
ICH: Technical Requirements for Registration of Pharmaceuticals for Human Use: Q9- Quality Risk Managment. 2004
10
ICH: Technical Requirements for Registration of Pharmaceuticals for Human Use: Q3C Impurities: Guideline for Resi dual Solvents. 2018
11
PIC/S: Validation Masterplan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation. 2007
12
US FOOD AND DRUG ADMINISTRAI ON: Code of Federal Regulations Title 21, PART 184 DIRECT FOOD SUBSTANCES AFFIRMED AS GENE RALLY RECOGNIZED AS SAFE.– URL https://www.accessdata.fda.gov/ scripts/cdrh/cfdocs/cfcfr/ CFRSearch.cfm?CFRPart=184