1 Einleitung
Eine schnelle, zuverlässige und deshalb häufig verwendete Methode zur Sterilisation von wässrigen Zubereitungen im Endbehältnis ist die Dampfsterilisation in einem Autoklav. Wir betrachten beispielhaft Injektions- und Infusinsflaschen als die am meisten verwendeten Packmittel für Parenteralia sowie Fertigspritzen.
Die Primärpackmittel werden geeignet vorbehandelt, z.B. werden Flaschen aus Glas zunächst gewaschen, sterilisiert und entpyrogenisiert. Anschließend werden sie mit der wässrigen Zubereitung befüllt und mit Stopfen verschlossen. Die Stopfen von Flaschen werden durch Bördelkappen fixiert. Bei Fertigspritzen wird der Stopfen nur eingepresst, da dieser bei der Applikation verschoben werden muss. Mit geeigneten Aufnahmevorrichtungen und Beschickungswagen werden die Behältnisse in einen Sterilisator gestellt und mit Dampf bei 121 C und 2 bar (0,2 MPa) sterilisiert.
In Fachbüchern der Pharmazeutischen Technologie, z.B. [2, 12], wird auf die Besonderheit der Sterilisation von Flüssigkeiten in geschlossenen Behältnissen hingewiesen. In diesen baut sich ein Binnendruck auf, welcher zum Bersten führen kann. Von Voigt [11] wurde eine Tabelle für den Binnendruck und den resultierenden Differenzdruck zur Sterilisationskammer bei verschiedenen Füllgraden vorgestellt, jedoch kein Rechenweg angegeben. Bei der Anwendung des Dampf-Luftgemisch- Verfahrens muss dieser Differenzdruck, auch Stützdruck genannt, mittels eingepresster, steriler Druckluft ausgeglichen werden, so dass eine genaue Berechnung in Abhängigkeit vom Füllgrad und weiteren Einflüssen wünschenswert ist. Einige der Mechanismen der Binnendruckerhöhung wurden für verschiedene Formen von Primärverpackungen bereits beschrieben, siehe [3], [10], [7] und [4].
An erster Stelle ist hier die grundlegende Arbeit von Beck [3] zu nennen, der vor mehr als 35 Jahren eine Berechnungsgleichung für den Druck in verschlossenen Flaschen beim Erhitzen im Autoklav entwickelt hat. Seine Veröffentlichung ist leider durch eine Vielzahl von Druckfehlern entstellt worden. Auch in seiner einige Monate später abgedruckten Fehlerberichtigung sind nicht alle Druckfehler erkannt und beseitigt worden. Diese immer noch fehlerhafte Gleichung wurde später in der Arbeit von Joyce und Lorenz [7] erneut abgedruckt. Das Ziel dieses Berichts besteht deshalb darin, die Grundlagen der von Beck aufgestellten Druckgleichung zu erläutern, ihre korrekte Form abzuleiten und ihre Anwendung zu demonstrieren.
2 Die Mechanismen der Druckerhöhung
Der Flüssigkeitsspiegel unterteilt den Innenraum des Behältnisses in den Kopfraum mit der Gasphase und den Produktraum mit der Flüssigkeitsphase. Die Gasphase besteht aus einem Luft-Wasserdampf-Gemisch, das nach dem Verschließen des Behältnisses rasch in den Sättigungszustand übergeht. Die damit einhergehende Änderung des Flüssigkeitsspiegels durch Masseabnahme der Flüssigkeit soll hier vernachlässigt werden.
Arzneimittel als wässrige Lösungen kleiner Moleküle, z.B. isotonische Kochsalzlösung, enthalten gelöste Stoffe nur in sehr geringen Konzentrationen. Große Moleküle, z.B. Proteine als Wirkstoffe oder polymere Gerüstbildner in Gelen, bewirken auch bei hohen Konzentrationen nur eine sehr geringe Erniedrigung des Dampfdrucks. Daher wird die flüssige Phase zur Vereinfachung wie reines Wasser betrachtet. Das Volumen des gesamten Innenraums unseres Behältnis Modells wird mit V bezeichnet, das Volumen des Kopfraumes mit Vg (Index g = Gasphase) und das Volumen des Produktraumes mit Vf (Index f = Flüssigkeitsphase).$$V = V_g + V_f$$
Der Anteil des Kopfraumvolumens am Gesamt-
volumen beträgt $$y = V g /V$$ und der Anteil des
Produktraumes (Füllgrad) entspricht:$$1 – y = V_f/V$$
Der Anfangszustand (Index 1) wird durch die Temperatur T1 , den Druck p1 und den Volumenanteil y1 festgelegt. Die Erwärmung des Behältnisses soll so langsam erfolgen, dass bei jedem neuen Zustand sich ein thermodynamisches Gleichgewicht (Index 2) ausbildet. Insbesondere sollen instationäre Wärmeleitungs- und Wärmespeicherungseffekte vernachlässigt werden. Sind y1 und y2 bekannt, kann die Änderung des Volumenanteils mit Hilfe eines so genannten Kompressionsfaktors f y ausgedrückt werden:
$$f_y = \frac{y_1}{y_2}$$
Die folgenden Effekte bewirken eine Druckerhöhung im Behältnis:
1. Das Behältnis dehnt sich bei der Temperaturerhöhung aus und sein Volumen nimmt von V 1 auf V 2 zu. Dadurch wird der Druck geringfügig abnehmen.
2. Ein Teil der Flüssigkeit verdampft, bis ein neuer Sättigungszustand erreicht ist. Der Partialdruck des Wasserdampfs steigt auf den Wert des Sättigungsdampfdrucks pDS,2 .
3. Der Partialdruck der Luft nimmt gemäß dem idealen Gasgesetz mit der Temperatur zu.
4. Die Flüssigkeitsphase dehnt sich infolge der Temperaturänderung aus, d.h. der Flüssigkeitsspiegel steigt und komprimiert im Gegenzug die Gasphase (y2 < y 1 ). Die Flüssigkeit selbst wird als inkompressibel angenommen, d.h. die Kompression des Wassers infolge des Druckanstiegs kann im Gegensatz zur Gasphase vernachlässigt werden.
5. Ein Teil der Luft, die im Ausgangszustand in der Flüssigkeit gelöst waren, werden bei steigender Temperatur aufgrund der sinkenden Löslichkeit in den Kopfraum ausgasen und damit einen weiteren Beitrag zur Druckerhöhung im Behältnis leisten. Ab einem bestimmten Partialdruck der Luft kehrt sich dieser Effekt um und die Luft wird mit steigendem Druck wieder in der Flüssigkeit gelöst.
3 Ausdehnung des Behältnisses
Wenn wir das Behältnis auf einen allseits geschlossenen Zylinder reduzieren, kann das Volumen unseres Behältnismodells wie folgt beschrieben werden:
$$V = \frac{\pi}{4} D^2 L,$$
wobei D = der innere Durchmesser und L = die innere Länge des Zylinders sind. Bei der Erwärmung des Zylinders wird sein Volumen vergrößert:
$$\frac{dV}{V} = 2 \cdot \frac{dD}{D} + \frac{dL}{L}$$
Das Gesetz der linearen Ausdehnung lautet wie folgt:$$\frac{dD}{D} = \frac{dL}{L} = \alpha dT$$
Damit erhalten wir für die relative Volumenver- größerung:$$\frac{dV}{V} = 3\alpha dT$$
Bei einer endlichen Temperaturerhöhung dT = T₂ – T₁ erhalten wir
$$dV_1 = 3\alpha dT V_1 $$
$$V_2 = V_1 + dV_1 $$
$$V_2 = V_1 [1 + 3\alpha(T_2 – T_1)],$$
wobei der lineare Ausdehnungskoeffizient α hier als konstant angenommen wird. Mit dem Volumen des Behältnisses vor und nach der Zustandsänderung soll hier der Kompressionsfaktor fα definiert werden.
$$f_\alpha = \frac{V_1}{V_2}$$
4 Der Druck in der Gasphase
Der Gesamtdruck in der Gasphase ist nach dem Gesetz von Dalton gleich der Summe der Partialdrücke. Die Luft ist gesättigt, so dass der Partialdruck des Wasserdampfs dem Sättigungsdampfdruck entspricht.
$$p_1 = p_{L,g,1} + p_{W,g,1} = p_{L,g,1} + p_{DS,1} $$
$$p_2 = p_{L,g,2} + p_{W,g,2} = p_{L,g,2} + p_{DS,2}$$
Der Partialdruck der Luft wird nun mit dem Gesamtdruck und dem Sättigungsdampfdruck ausgedrückt.
$$p_{L,g,1} = p_1 – p_{DS,1}$$
$$p_{L,g,2} = p_2 – p_{DS,2}$$
Der unbekannte Partialdruck der Luft im Zu- stand 2 kann wegen p/T=konstant leicht ermittelt werden.
$$p_{L,g,2} = p_{L,g,1} \frac{T₂}{T_1} = (p_1 – p_{DS,1}) \frac{T_2}{T_1}$$
Somit erhält man den Druck bei einer isochoren Zustandsänderung mit einem konstanten Volumen und ohne Änderung der Massen in der Gasphase.
$$p_2 = (p_1 – p_{DS,1}) \frac{T_2}{T_1} + p_{DS,2}$$
An dieser Stelle soll als Hilfsgröße der thermische Kompressionsfaktor fᴛ eingeführt werden, welcher dimensionslos den Temperatureinfluss ausdrückt
$$f_T = \frac{T_2}{T_1}$$
5 Dampfdruck und Dichte von Wasser
5.1 Sättigungsdampfdruck des Wassers
Nach der Internationalen Wasserdampftafel von 1997 [1] wird der Sättigungsdampfdruck pDS in der Einheit MPa wie folgt berechnet:
$$
\frac{p_{DS}}{p^\ast} = \left[ \frac{2C}{B + \sqrt{-B^2 – 4AC}} \right]^4
$$
mit p* = 1 MPa, eine Hilfsgröße zur Erzeugung einer dimensionslosen Druckgröße. Die anderen Hilfsgrößen haben die folgende Bedeutung:
$$A = n_0 \vartheta^2 + n_1 \vartheta + n_2 $$
$$B = n_3 \vartheta^2 + n_4 \vartheta + n_5 $$
$$C = n_6 \vartheta^2 + n_7 \vartheta + n_8$$
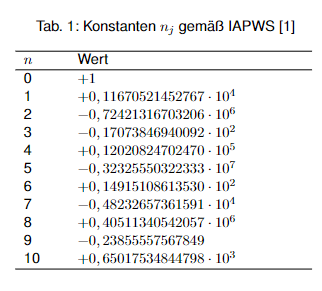
und die darin verwendete dimensionslose Temperatur ϑ wird wie folgt definiert:
$$\vartheta = \frac{T_s}{T^\ast} + \frac{n_9}{\frac{T_s}{T^\ast} + n_{10}}$$
mit Ts= thermodynamische Temperatur am Sättigungspunkt und T* = 1 K, einer Hilfsgröße zur Erzeugung einer dimensionslosen Temperatur. Die verwendeten Konstanten nj sind in Tab. 1 aufgelistet.
5.2 Die Dichte des flüssigen Wassers
Bei Temperaturerhöhung dehnt sich flüssiges Wasser aus und seine Dichte nimmt ab, was zum Druckanstieg im Behältnis führt. Die Dichte Im Sättigungszustand kann gemäß dem Stoffdatenblatt 11 der PTB [9] wie folgt mit den Konstanten aus Tab. 2 berechnet werden.
$$\varrho_r = 1 + \sum_{i=2}^6 A_i (1 – T_r)^{a_i}$$
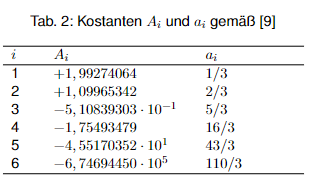
mit T r = reduzierte Temperatur und %r = reduzierte Dichte. Die reduzierten Größen sind mit Hilfe der kritischen Größen Tc und ϱc wie folgt definiert:
$$T_r = \frac{T}{T_c} \quad \text{mit} \quad T_c = 647,096 \, \text{K}$$
$$\varrho_r = \frac{\varrho}{\varrho_c} \quad \text{mit} \quad \varrho_c = 322 \, \text{kg/m}^3$$
Die so berechnete Dichte ϱ geht später in die temperaturabhängige Massenbilanz des Wassers als ϱw,f ein.
6 Löslichkeit von Luft in Wasser
6.1 Spezifische Gaskonstanten
Für die spätere Ableitung der Druckgleichung und ihrer Berechnung werden die Molmassen und die spezifischen Gaskonstanten der Luft und des Wassers benötigt. Die Molmassen der Luft und des Wassers weisen nach IUPAC-97 [6] folgende Werte auf:
$$M_{L} = 28,963 \text{ g/mol} $$
$$M_{W} = 18,015257 \text{ g/mol}$$
Die molare Gaskonstante wird nach CODATA 2018 [8] mit Rₘ = 8,314472 J/molK angegeben. Damit können die spezifischen Gaskon- stanten der Luft und des Wassers ermittelt werden:
$$ R_L = R_m / M_L = 287,072196 \text{ J/(kgK)} $$
$$ R_W = R_m / M_W = 461,523918 \text{ J/(kgK)} $$
6.2 Gesetz von Henry
In ideal verdünnten Lösungen gilt das Gesetz von Henry, das auf die Materialpaarung Luft- Wasser angewendet wie folgt lautet: der Molenbruch xL,h (Index h für gelöst) der im Wasser gelösten Luft ist proportional dem Partialdruck pL,g , den die Luft oberhalb des Wasserspiegels ausübt.
$$ p_{L,g} = H x_{L,h} $$
Die Proportionalitätskonstante H wird Henry- Konstante genannt. Sie ist eine Materialkonstante, die im Allgemeinen von der Materialpaarung Gas-Flüssigkeit und von der Temperatur abhängt. Der Molenbruch der gelösten Luft wird wie folgt ausgedrückt:
$$x_{L,h} = \frac{n_{L,h}}{n_{L,h} + n_W}$$
mit n L,h = Stoffmenge der Luft in mol, die im Wasser gelöst ist, und n w = Stoffmenge des reinen Wassers in mol, in dem die Luft gelöst wird. Weil die Stoffmenge der gelösten Luft sehr viel kleiner ist als die des Wassers, kann man Gl. 5 wie folgt vereinfachen:
$$ x_{L,h} \approx \frac{n_{L,h}}{n_W} \quad \text{wegen} \quad n_{L,h} \ll n_W $$
Für die Stoffmenge der gelösten Luft erhalten wir damit den folgenden Ausdruck:
$$ n_{L,h} = \frac{n_W}{H} \cdot p_{L,g} \quad (6) $$
Für die spätere Massenbilanz ersetzen wir in Gl. 6 die Stoffmengen durch die Massen mit Hilfe der folgenden Gleichungen:
$$m_{L,h} = n_{L,h} M_L $$
$$m_W = n_{W,f} M_W$$
und damit den Ausdruck für die Masse der gelösten Luft:
$$ m_{L,h} = m_W \frac{M_L}{M_W} p_{L,g} \frac{1}{H} $$
$$ = \varrho_W (1-y)V \frac{M_L}{M_W} (p – p_{DS}) \frac{1}{H} $$
6.3 Die Temperaturabhängigkeit der Henry-Konstante
Die Henry-Konstante H für die Stoffpaarung Luft-Wasser ist abhängig von der Temperatur: je höher die Temperatur des Wassers, desto weniger Luft kann im Wasser gelöst werden. Bei sehr hohen Drücken ist H auch abhängig vom Druck. Diese Druckabhängigkeit kann bei den hier in Frage stehenden Drücken vernachlässigt werden.
In Dorsey [5] (Table 232/III, p.: 539) wurden die Werte für 1/H im Temperaturbereich 0 C t 100 C tabelliert. Die Werte wurden mit der Einheit 10-9 /mm Hg angegeben. Daraus wurde hier der Kehrwert H gebildet und auf die Einheit atm umgerechnet, um eine Vergleichbarkeit mit der Formulierung von Beck herzu- stellen. Anschließend wurden die Werte auf die Einheit Pa umgerechnet, um sie SI-konform in der Druckgleichung verwenden zu können.
6.4 Beispiel
Bei 20 C findet man in der Dorsey-Tafel den
Wert: 1/H=19,82 10⁻⁹/mm Hg. Die Umrechnung in die SI-Einheit lautet wie folgt:
$$ \frac{1}{H} = 19,82 \frac{10^{-9}}{mmHg} \frac{760 \text{ mmHg}}{101,325 \text{ Pa}} $$
$$ = 1,4866 \cdot 10^{-1} / \text{Pa} $$
Damit erhalten wir bei 20 C die Henry- Konstante mit H20 = 6,7267 10⁹ Pa = 6,6387 10⁴ atm. Bei Beck [3] findet man die folgende Formulie- rung der Henry-Konstante:
$$ \frac{H}{H_{20}} = 1,627 – \frac{223}{T} \exp \left[ -\left( \frac{T – 273,2}{45} \right)^2 \right] \quad (7) $$
wobei H₂O den Wert von 6,64 10⁴ atm annimmt, also in guter Übereinstimmung mit dem Tafel- wert von Dorsey steht. Die Temperatur T wird gemäß [3] in Gl. 7 in K eingesetzt, um H in atm zu erhalten. Die Gleichung von Beck lie- fert für t > 20 C gute Näherungswerte im Ver- gleich zu den Tafelwerten von Dorsey. Unter- halb von 20 C ist die Approximationsgleichung von Beck zu ungenau. Deshalb wurde hier eine eigene Approximationsgleichung (Gl. 8) für die Temperaturabhängigkeit der Henry-Konstante entwickelt, um in der Wahl des Bezugspunktes frei zu bleiben und eine höhere Genauigkeit zu erhalten. Die Konstanten bₖ sind in Tab. 3 auf- gelistet.
$$ \frac{H_0}{H} = \exp \left[ -\left( \frac{t}{65} \right)^2 \right] – \sum_{k=0}^5 b_k \cdot t^k \quad (8) $$
Die Gl. 8 ist im Bereich 0°C ≤ t ≤ 140°C gültig mit Hₒ = Hₜ = 0°C = 4,3698 · 10⁹ Pa bzw. = 4,3698 · 10⁴ atm. Zur Abstraktion des Einflusses der Löslichkeit soll wieder ein Kompressionsfaktor eingeführt werden. Die Formulierung des Kompressions- faktors fₕ (siehe Gl. 29) setzt die Umstellung Tab. 3: Konstanten der Approximationsgleichung 8

der Massenbilanzen voraus, welche im nächsten Abschnitt vorgenommen werden.
7 Die Druckgleichung
7.1 Vorgehensweise
Ziel ist es, einen expliziten Ausdruck für die Berechnung des Drucks p₂ aufzustellen. Die Entwicklung der Druckgleichung erfolgt in vier Schritten:
- Aufstellung der Massenbilanz des Wassers und Umstellung nach dem Volumenanteil y₂ der Gasphase infolge der Temperaturänderung von T₁ nach T₂ ,
- Aufstellung der Massenbilanz der Luft für beide Zustände,
- Kombination der Massenbilanzen zur Formulierung einer impliziten Gleichung (Gl. 26) für den Druck p₂ ,
- Umstellung in einen expliziten Ausdruck für den Druck p₂ (Gl. 28).
7.2 Bilanz der Wassermassen
Die Masse des Wassers im Produktraum (Masse der flüssigen Phase) und die Masse des Wasserdampfs im Kopfraum müssen vor und nach der Zustandsänderung gleich sein.
$$ \left. m_{w,f} + m_{w,g} \right|_{1} = \left. m_{w,f} + m_{w,g} \right|_{2} \quad (9) $$
Die Masse des flüssigen Wassers ohne gelöste Bestandteile im Zustand 1 wird beschrieben durch
$$m_{w,f,1} = \varrho_{w,f,1} V_{w,f,1} = \varrho_{w,f,1} (1-y_1)V_1 \quad (10)$$
und die Masse des flüssigen Wassers im Zu- stand 2 entsprechend mit
$$m_{w,f,2} = \varrho_{w,f,2} V_{f,2} = \varrho_{w,f,2} (1-y_2)V_2. \quad (11)$$
Zur besseren Übersicht soll der Index „D“ für den gasförmigen Wasserdampf den Index „w,g“ sowie der Index „W“ für das flüssige Was- ser den Index „w,f“ ab jetzt ersetzen. Die Mas- se des Wasserdampfs im Zustand 1 wird be- schrieben durch
$$m_{D,1} = \varrho_{D,1} V_{g,1} = \varrho_{D,1} y_1 V_1$$
$$= \frac{p_{DS,1}}{R_D T_1} y_1 V_1 \quad (12)$$
und die Masse des Wasserdampfs im Zustand 2 entsprechend mit
$$ m_{D,2} = \varrho_{D,2} V_{g,2} = \varrho_{D,2} y_2 V_2 $$
$$ = \frac{p_{DS,2}}{R_D T_2} y_2 V_2. \quad (13) $$
Werden die Gleichungen 10 bis 13 in Gl. 9 ein- gesetzt, ergibt sich für den Volumenanteil des Kopfraums im Zustand 2 der folgende Zusammenhang:
$$ y_2 = \frac{1 – \frac{\varrho_{w,1}}{\varrho_{w,2}} \left[ 1 – y_1 \left( 1 – \frac{\varrho_{D,1}}{\varrho_{w,1}} \right) \right] \frac{V_1}{V_2}}{1 – \frac{\varrho_{D,2}}{\varrho_{w,2}}} \quad (14) $$
Die Dichte des Dampfes ist sehr viel kleiner ist als die Dichte des flüssigen Wassers
$$ \left. \frac{\varrho_D}{\varrho_W} \right|_1 \quad \text{bzw.} \quad \left. \frac{\varrho_D}{\varrho_W} \right|_2 \ll 1 \quad (15) $$
und die Gl. 14 kann vereinfacht werden:
$$ y_2 \simeq 1 – \frac{\varrho_{W,1}}{\varrho_{W,2}} (1 – y_1) \frac{V_1}{V_2} \quad (16) $$
Wird nun die Volumenausdehnung des Behält- nisses gemäß Gl. 2 berücksichtigt, kann die Gl. 16 wie folgt geschrieben werden:
$$ y_2 \simeq \frac{\varrho_{w,1}}{\varrho_{w,2}} \left[ \frac{1 – y_1}{1 + 3 \alpha (T_2 – T_1)} \right] \quad (17) $$
7.3 Bilanz der Luftmassen
Die Masse der Luft im Kopfraum und die im Wasser gelöste Luftmasse muss vor und nach der Zustandsänderung ebenfalls gleich sein.
$$ m_{L,g} + m_{L,h} \bigg|_1 = m_{L,g} + m_{L,h} \bigg|_2 $$
Die Masse des Luftanteils in der Gasphase des Kopfraums im Zustand 1 wird beschrieben mit
$$ m_{L,g,1} = \varrho_{L,g,1} \, y_1 \, V_1 \quad (19) $$
Gleichzeitig gilt mit der idealen Gasgleichung
$$pV = mRT$ bzw. $\varrho_{L,g,1} = p_{L,1} / (R_L T_1)$$
$$ m_{L,g,1} = \frac{p_{L,1}}{R_L T_1} y_1 V_1 \quad (20) $$
$$ = \frac{p_{L,1} M_L}{R_M T_1} y_1 V_1 $$
bzw.
$$ m_{L,g,1} = (p_1 – p_{DS,1}) \frac{M_L}{R_M T_1} y_1 V_1 \quad (21) $$
und im Zustand 2 gilt dementsprechend
$$m_{L,g,2} = \varrho_{L,2} y_{2} V_{2} \quad (22)$$
$$ \frac{p_{L,2}}{R_{L} T_{2}} y_{2} V_{2} = (p_{2} – p_{DS,2}) \frac{M_{L} y_{2} V_{2}}{R_{M} T_{2}} \quad (23) $$
Für die Masse der Luft, welche in der Flüssigkeit gelöst ist, gilt dann
$$m_{L,f,1} = \varrho_{w,1} (1 – y_{1}) V_{1} (p_{1} – p_{DS,1}) \frac{M_{L}}{M_{W}} \frac{1}{H_{1}} \quad (24)$$
und
$$m_{L,f,2} = \varrho_{w,2} (1 – y_{2}) V_{2} (p_{2} – p_{DS,2}) \frac{M_{L}}{M_{W}} \frac{1}{H_{2}} \quad (25)$$
7.4 Implizite Gleichung nach Beck
Die Gleichungen 20 bis 25 werden jetzt in die
Bilanzgleichung 18 eingesetzt. Es ergibt sich
ein impliziter Ausdruck für den Druck p₂ .
$$ \frac{p_{2} – p_{DS,2}}{p_{1} – p_{DS,1}} = \frac{\frac{y_{1}}{R_{m}T_{1}} + \varrho_{w,1} \frac{(1 – y_{1})}{M_{W}H_{1}}}{\frac{y_{2}}{R_{m}T_{2}} + \varrho_{w,2} \frac{(1 – y_{2})}{M_{W}H_{2}}} \frac{V_{1}}{V_{2}} \quad (26) $$
Durch Umstellung ergibt sich die von Beck ver- öffentlichte Gleichung, die nach der Einarbei- tung aller von ihm mitgeteilten und weiteren an- deren Korrekturen wie folgt lautet:
$$ \frac{p_{2} – p_{DS,2}}{p_{1} – p_{DS,1}} = \frac{1 + \frac{y_{1}}{1 – y_{1}} \frac{M_{W}H_{1}}{\varrho_{w,1}R_{m}T_{1}}}{\frac{M_{W}H_{1}}{\varrho_{w,1}R_{m}T_{2}} \left[ \frac{1 + 3\alpha(T_{2} – T_{1})}{1 – y_{1}} – \frac{\varrho_{w,1}}{\varrho_{w,2}} \right] + \frac{H_{1}}{H_{2}}} \quad (27) $$
In diesem Ausdruck ist der Volumenanteil y 2 gemäß Gl. 17 vollständig eingearbeitet, so dass auf der rechten Seite von Gl. 27 nur noch bekannte, vorgegebene bzw. berechenbare Größen erscheinen.
7.5 Explizite Gleichung für den Druck
Durch Verzicht auf die Einarbeitung von Gl. 17, jedoch mit Hilfe der Einführung des Terms fₕ , welcher y₂ ergibt sich p₂ wie folgt:
$$p_2 = p_{DS,2} + (p_1 – p_{DS,1}) \frac{V_1}{V_2} \frac{T_2}{T_1} \frac{y_1}{y_2} f_H \quad (28)$$
Der Term fₕ soll dabei als Kompressionsfaktor der Löslichkeit interpretiert werden.
$$f_{H} = \frac{1 + \frac{1 – y_1}{y_1} \frac{\rho_{w,2} R_m T_1}{M_W} \frac{1}{H_1}}{1 + \frac{1 – y_2}{y_2} \frac{\rho_{w,2} R_m T_2}{M_W} \frac{1}{H_2}} \quad (29)$$
Weiter werden alle durch Umstellung isolierten Verhältnisse der Volumenausdehnung, der Temperaturerhöhung und des Kopfraumvolumenanteils durch ihren entsprechenden Kompressionsfaktor ersetzt. Durch geeignete Umstellung erhält man die folgende Gleichung:
$$P_2 = P_{DS,2} + (P_1 – P_{DS,1}) \frac{V_1}{V_2} \frac{T_2}{T_1} \frac{y_1}{y_2} f_H \quad (30)$$
Es soll angemerkt werden, dass diese Glei- chung identische Werte wie die Gl. 27 von Beck liefert. Der Vorteil der Gl. 30 besteht darin, dass die Kompressionsmechanismen mit Hilfe ihrer Kompressionsfaktoren dimensionslos ausgedrückt werden können, was im nächsten Abschnitt erläutert werden soll.
8 Quantifizierung der Einflüsse
8.1 Kompressionsfaktoren
Die bisher abgeleiteten Kompressionsfaktoren Gl. 1, Gl. 3, Gl. 4 und Gl. 29 lassen sich nun zu einem dimensionslosen Gesamt Kompressionsfaktor f ges zusammenfassen.
$$ f_{\text{ges}} = f_{\alpha} f_{T} f_{y} f_{H} $$
Die Druckgleichung 30 vereinfacht sich dann weiter zu:
$$ p_2 = p_{DS,2} + (p_1 – p_{DS,1}) f_{\text{ges}} $$
$$ = p_{DS,2} + p_{L,1} f_{\text{ges}} $$
Der Enddruck p₂ im Behältnis steigt bis zum Sättigungsdampfdruck pDS,₂ bei der Endtemperatur Z₂ plus dem Partialdruck pL,₁ der Luft im Kopfraum bei Anfangstemperatur multipliziert mit dem Kompressionsfaktor fges .
8.2 Einfluss der Wärmeausdehnung des Behältnisses
Mit dem Kompressionsfaktor f V aus Gl. 3 können wir den Einfluss der Wärmeausdehnung des Behältnisses auf den Druck als Verhältnis der Volumina im Zustand 1 zu Zustand 2 beschreiben. Aus T₂ > T₁ folgt V₁ /V₂ < 1, d.h. die Wärmeausdehnung des Behältnisses wirkt druckentlastend. Dieser Effekt ist stark abhängig von der Wahl des Behältnismaterials. In Abb. 1 ist fα als Funktion der Temperatur für die Werkstoffe Glas und High-Density Polyethylen (HD-PE) aufgetragen. Bei Annahme eines linearen Ausdehnungskoeffizienten von α = 5 · 10⁻⁶ 1/K für Glas und α= 159 · 10⁻⁶ 1/K für HD-PE beträgt der Kompressionsfaktor f ↵ = 0,995 bzw. fα = 0,955. Die Glasbehältnisse verhalten sich nahezu wie starre Körper, während bei Behältnissen aus HD-PE eine Ausdehnung von fast 5 % erfahren.
8.3 Einfluss der Erwärmung
Der zweite Kompressionsfaktor fₜ ergibt sich aus dem Verhältnis der absoluten Temperaturen vor und nach der Zustandsänderung (sie- he Gl. 4). Herrscht eine Raumtemperatur von 20 C während der Abfüllung, so erhält man fₜ = 1,34. Die Erwärmung allein bewirkt eine Druckerhöhung um ca. 35 %. Aus der Abb. 2 kann dieser Wert für andere Abfülltemperaturen bezogen auf eine Sterilisationstemperatur von 121°C entnommen werde.
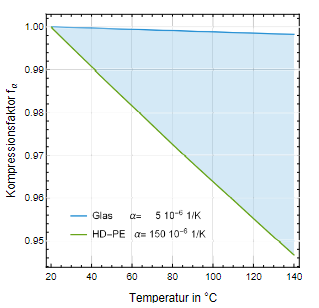
Abb. 1: Druckentlastung infolge der Ausdehnung des Behältnisses
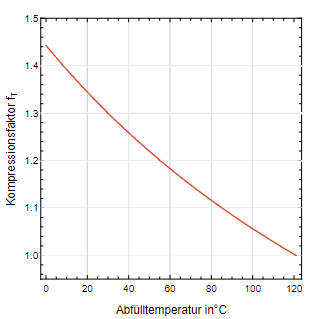
Abb. 2: Kompressionsfaktor fₜ in Abhängigkeit von der Abfülltemperatur
8.4 Einfluss des Kopfraums
Der dritte Faktor fy (siehe Gl. 1) repräsentiert die Verringerung des Kopfraums und ist der einflussreichste. Die Abb. 3 veranschaulicht die Verkleinerung des Kopfraumvolumenanteils y als Funktion der Temperatur. Für den Werkstoff Glas erkennt man, dass ein anfänglicher Kopfraumanteil von 10 % bei Erreichen der Sterilisationstemperatur auf ca. 5 % komprimiert wird. Dementsprechend beträgt der Kompressionsfaktor f y = 2,1 (Abb. 4), d.h. dass sich der Innendruck verdoppelt. Wird der Volumenanteil
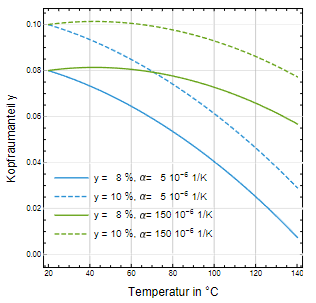
des Kopfraums auf 8 % herabgesetzt, steigt der Kompressionsfaktor auf f y = 2,9, d.h. der Innendruck verdreifacht sich. Betrachtet man dagegen HD-PE als Behältnismaterial, so fällt die Druckerhöhung deutlich niedriger aus (sie- he Abb. 3 und 4).
8.5 Einfluss der Löslichkeit der Luft
In Abb. 5 ist der Faktor fₕ als Funktion der Temperatur dargestellt. Es wird wieder beispielhaft ein Kopfraumanteil von 8 % oder 10 % angenommen. Zu Anfang wird die im Wasser gelöste Luft noch ausgetrieben und bewirkt damit eine geringfügige Druckerhöhung mit einem Maximum bei ca. 40 C. Steigt die Temperatur über 60 C, wird der Partialdruck der Luft so groß, dass die Luft wieder in das Wasser eingetrieben wird und damit eine Druckentlastung im Behältnis bewirkt.
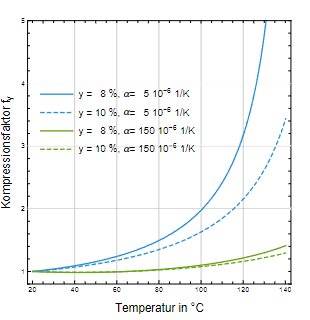
Abb. 4: Kompressionsfaktor des Kopfraumvolumenanteils fy
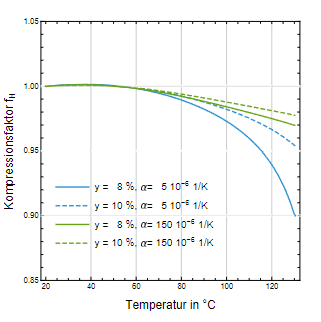
Abb. 5: Kompressionsfaktor der Löslichkeit von Luft in Wasser
Der Einfluss auf den Druck ist jedoch insgesamt gering, wie der Vergleich mit und ohne Gaslöslichkeit für einen Kopfraumanteil von 8 % oder 10 % in Abb. 6 zeigt.
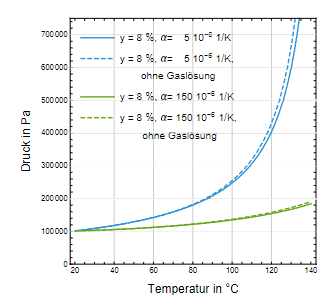
Abb. 6: Druckentlastung als Folge der Löslichkeit von Luft in Wasser
Es stellt sich jedoch die Frage, ob die Luft in der relativ kurzen Aufheizphase tatsächlich gelöst wird, d.h. ein thermodynamisches Gleichgewicht erreicht wird, wie die Gl. 29 dies voraussetzt. Bei großvolumigen Behältnis- sen, z.B. Röntgenkontrastmitteln in Flaschen, besteht eine gewisse Distanz zwischen Außenfläche und dem Kern der Flüssigkeit auf Grund der Wärmeleitung. Der Temperaturgradient resultiert in einer Konvektionsbewegung der Flüssigkeit, wodurch die Grenze zur Gasphase häufig erneuert wird und die Diffusion der Luft in die flüssige Phase begünstigt wird. Bei kleinvolumigen Behältnissen, z.B. Injektionslösungen in Fertigspritzen, ist der Temperaturgradient kleiner und die resultierende Konvektion entsprechend schwächer. Daher ist eine langsamerer Lösungsvorgang der Luft zu erwarten, d.h. ein Gleichgewicht wird verzögert erreicht.
8.6 Der Stützdruck
Der Druck im Behältnis wird nun unter Berücksichtigung der vorgestellten Druckmechanismen ermittelt. In der Abb. 7 ist der absolute Druck im Behältnis als Funktion der Temperatur für Glas und HD-PE dargestellt. Es wurde wieder von einem Kopfraumvolumenanteil von 8 % ausgegangen, jeweils mit und ohne Berücksichtigung der Löslichkeit der Luft im Wasser. Ist das Behältnis aus Glas, so herrscht bei einem Kopfraumanteil 8 % im Inneren bei 121 C ohne Berücksichtigung der Gaslöslichkeit ein Druck von ca. 4,2 bar und mit Berücksichtigung der Löslichkeit 4,0 bar. Stützdruck für Glasbehältnis ohne Gaslöslichkeit
$$ p_{\text{st}}(y_1 = 0{,}08) = p_2 – p_{DS}(121\,^\circ\mathrm{C}) $$
$$ = 4{,}2 – 2{,}0 = 2{,}2\,\text{bar} $$
Stützdruck für Glasbehältnis mit Gaslöslichkeit
$$ p_{\text{st}}(y_1 = 0{,}08) = p_2 – p_{DS}(121\,^\circ\mathrm{C}) $$
$$ = 4{,}0 – 2{,}0 = 2{,}0\,\text{bar} $$
Der Unterschied zwischen den beiden Fällen beträgt nur 0,2 bar. Wegen der Unsicherheit der Gleichgewichtseinstellung bezüglich der Gaslöslichkeit in der Aufheizphase sollte der größere Wert von p 2 = 4,2 bar bei der Festlegung des Stützdrucks berücksichtigt werden. Für HD-PE würde dagegen schon ein Stützdruck von ca. 0,6 bar genügen (Abb. 7), wobei die Gaslöslichkeit wegen des geringen Drucks kaum eine Rolle spielt.
Die Abb. 8 soll dies mit Hilfe des Gesamtkompressionsfaktors als Funktion von Temperatur und Kopfraumanteil verdeutlichen. Der Temperaturbereich wurde von 115 C bis 125 C gewählt, der Kopfraum zwischen 6 % bis 10 % variiert. Es ist gut zu erkennen, dass der Gesamt- Kompressionsfaktor bei Glas enorme Werte bei kleinem Kopfraum annimmt. Die damit einhergehende Druckerhöhung führt mit hoher Wahrscheinlichkeit zum Platzen der Behältnisse bzw. zum Verschieben der Stopfen von Spritzen. Anhand der deutlich flacheren Fläche für HD-PE wird deutlich, dass durch eine geeignete Wahl des Kopfraumanteils die Druckerhöhung bei der Sterilisation gut beherrschbar ist. Mit Hilfe der Abbildung 9 ist es schließlich möglich, die geeignete Kombination von Stützdruck und Kopfraumvolumenanteil für die vorgestellten Materialien Glas und HD-PE zu wählen.
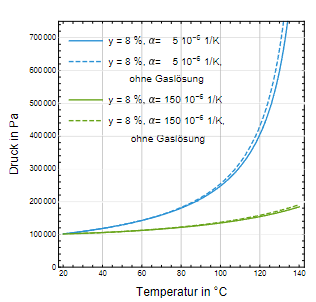
Abb. 7: Druckverlauf während der Sterilisation mit Kopfvolumenanteil 8 %, mit und ohne Gaslösung für Glas und HD-PE
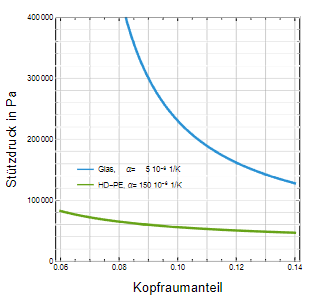
Abb. 9: Stützdruck in Abhängigkeit vom Kopfraumvolumenanteil für Glas und HD-PE
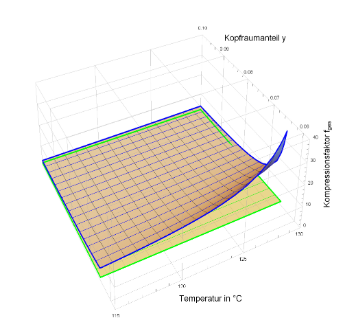
Abb. 8: Gesamt-Kompressionsfaktor für Glas (blaues Gitternetz) und HD-PE (grün)
9 Zusammenfassung
Die Zunahme des Druckes in geschlossenen Behältern während der Sterilisation durch Hit- ze kann auf vier von der Temperatur abhängige Kompressionsfaktoren abstrahiert werden, wo- bei die Reihenfolge die Stärke des Einflusses wiedergibt:
- fy Wahl des Kopfraumanteils
- fₜ Zuständsänderung durch Erwärmung
- fα Volumenänderung des Behälters
- fₕ Löslichkeit von Luft in Wasser
Die Kompressionsfaktoren fy und f T bilden die Grundlage für die Wahl der Prozessparameter. Entscheidend ist die Temperaturdifferenz während der Abfüllung und der Sterilisation. Je kleiner diese Differenz ausfällt, umso kleiner wird auch die Druckdifferenz zwischen dem Inneren des Behältnisses und der Sterilisationskammer. Wenn der Stützdruck aus technischen Randbedingungen rechnerisch zu hoch wird, muss der Kopfraum vergrößert werden. Gleiches gilt für die Verhinderung der Stopfenbewegung. Die Anpassung des Kopfraumanteils stellt daher die technisch einfachste Lösung dar.
Der Kompressionsfaktor fα ermöglicht es, das Material des Primärpackmittels zu berücksichtigen. Im Rahmen der Arzneimittelentwicklung kann damit das Primärpackmittel zugunsten von Materialien mit hohen thermischen Ausdehnungskoeffizienten gewählt werden, sofern diese keine Wechselwirkungen mit den Komponenten des Arzneimittels zeigen. Die Längenänderungen des Behälters bewirken eine Druckentlastung im Behälter während der Sterilisation und sind zu bevorzugen. Sind Behältnisse aus Glas erforderlich, muss der Prozess anhand der oben genannten Faktoren gestaltet werden.
Der Kompressionsfaktor fₕ zeigt grundsätzlich, dass eine Druckentlastung stattfindet, weil der steigende Partialdruck der Luft bewirkt, dass sich mehr Luft im Wasser löst. Dieser Diffusionsvorgang ist jedoch zeitabhängig, so dass das den Berechnungen zugrunde gelegte thermodynamische Gleichgewicht nur eine grobe Annahme ist. Wahrscheinlicher ist, dass dieses Gleichgewicht während des Sterilisationsprozesses nicht erreicht wird. Zusammen mit der Tatsache, dass dieser Einfluss quantitativ im Vergleich zu den anderen Kompressionsfaktoren kaum ins Gewicht fällt, sollte er bei vorwiegend wässrigen Lösungen vernachlässigbar sein.
In der Praxis wird man anhand der Temperaturen in den einzelnen Prozessphasen, wie z.B. Vorheizen, Sterilisieren und Kühlen, den jeweils erforderlichen Stützdruck berechnen und den Sterilisator dahingehend parametrieren. Behältnisse mit fixierten Verschlüssen wie Flaschen und Vials sind dabei unempfindlich gegen stufenweise Änderungen des Stützdruckes in sequentiellen Prozessphasen. Für Behältnisse mit beweglichen Verschlüssen wie Fertigspritzen sollte die Regelung des Stützdrucks möglichst kontinuierlich von der Innentemperatur der Behältnisse, sprich bei wässrigen Lösungen dem resultierenden Druck der Sattdampfkurve, abhängig gemacht werden. Anhand der hiervorgestellten Berechnungen konnte gezeigt werden, dass genügend Parameter zur Verfügung stehen, um in der Praxis einen Sterilisationszyklus für geschlossene Behältnisse wässriger Lösungen mit einem entsprechend ausgerüstetem Sterilisator zu realisieren.
Literatur
1
Revised Release on the IAPWS Industrial Formulation 1997 for the Thermodynamic Properties of Water and Steam. The International Association for the Properties of Water and Steam, Lucerne, 2007
2
BAUER, Kurt H. ; FRÖMMING, Karl-Heinz ; FÜHRER, Claus: Pharmazeutische Technologie. 5. Aufl. Georg Thieme Verlag, Stuttgart, 1997
3
BECK, Robert E.: Autoclaving of Solutions in Sealed Containers: Theoretical Pressure-Temperature Relationships, Pharmaceutical Manufacturing. (June 1985), S. pp.:18–23; Druckfehlerberichtigung im Heft September 1985, p. 12
4
BRYANT, Peter L.: Modeling of Parental Container Headspace Pressure. In: Technical Note in PDA Journal pf Pharmaceutical Science &Technology 52 (May-June 1998), Nr. 3, S. 123–128
5
DORSEY, Ernest N. ; 1957, February (Hrsg.): Properties of Ordinary Water Substance in all its Phases: Water-Vapor, Water, and all the Ices. N.Y. : Reinhold Publishing Corporation (3rd Printing)
6
IUPAC: Iupac Handbook 1996-97. Inter national Union of Pure Applied Chemistry, Portland, Or., 1997
7
JOYCE, Martin A. ; LORENZ, Jeffrey W.: In ternal Pressure of Sealed Containers During Autoclaving. In: Journal of Parental Science and Technology 44 (November December 1990), Nr. 6, S. 320–323
8
NATIONAL INSTITUTEOFSTANDARDS AND TECHNOLOGY, U.S. Department of Commerce: https://physics.nist.gov/cuu/ Constants/index.html.– Accessed : 2019-12-12
9
PHYSIKALISCH-TECHNISCHE BUNDES ANSTALT(PTB): PTB-Stoffdatenblätter, SDB 11: Wasser. Januar 1995
10
VENTURA, Dominic A. ; SHEAFFER, George E.: Unique Aspects of Steam Sterilization Validation of Disposable Syringe Components. In: Journal of Parental Science and Technology (November Dezember 1984), S. pp. 121–214
11
VOIGT, Rudolf: Pharmazeutische Technologie. 9. Aufl. Deutscher Apothekerverlag, Stuttgart, 2000
12
ZIMMERMANN, Ingfried: Pharmazeutische Technologie. Springer Verlag, Berlin, 1998
Wassergehalt und relative Feuchte
Das Gesetz von Dalton
Das Gesetz von Dalton bezieht sich auf Gasgemische. Dabei wird vorausgesetzt, dass die einzelnen Gase der Mischung nach dem Idealgasgesetz behandelt werden dürfen. Feuchte Luft bzw. feuchter Stickstoff sind solche typischen Vertreter solcher Idealgasgemische. Sie setzten sich zusammen aus reiner trockener Luft bzw. reinem trockenem Stickstoff und Wasserdampf, der als gasförmiger Aggregatzustand des Wassers ebenfalls nach dem Idealgasgesetz agiert. Nach Dalton füllt jedes einzelne Gas der Gasmischung das gesamte zur Verfügung stehende Volumen V aus und übt dabei einen sog. Partialdruck aus. Die Summe der Partialdrücke ergibt den Gesamt druck p, der mit einem Manometer an einer kleinen Bohrung in der Gefäßwand gemessen werden kann:

wobei pₗ = Partialdruck der Luft und pd = Partial druck des Wasserdampfes ist.
Der Wassergehalt
Der Wassergehalt x wird wie folgt definiert:

mit md= Masse des Dampfes und ml = Masse der trockene Luft. Der Wassergehalt als Konzentrationsgröße bezieht sich auf die Masse der reinen trockenen Luft, sehr ähnlich wie bei der Definition der Molalität. Man wendet nun das Idealgasgesetz auf beide Komponenten an:
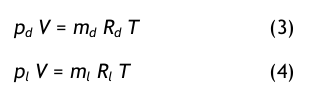
mit den speziellen Gaskonstanten für den Dampf, Rd, bzw. für die Luft Rl. Aus diesen bei den Gleichungen erhält man
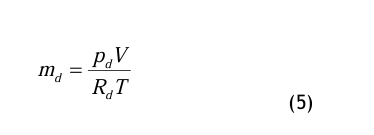
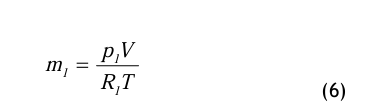
Die Gln.(5) und (6) werden in die Definitionsgleichung (2) für den Wassergehalt eingesetzt und wie folgt umgeformt:
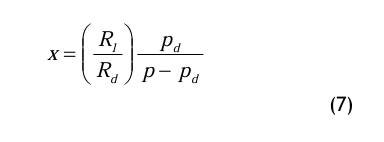
Die relative Feuchte
An dieser Stelle wird die relative Feuchte φ ein geführt und wie folgt definiert:
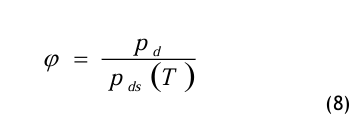
Die relative Feuchte ist eine Verhältniszahl, wobei der Partialdampfdruck pd auf den Sättigungsdampfdruck pds (T) bezogen wird. Der Dampfdruck im Sättigungszustand pds ist nur von der Temperatur T abhängig. Die Gl.(8) wird in die Gl.(7) eingesetzt:
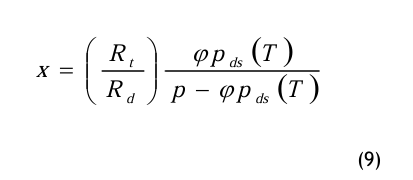
Die speziellen Gaskonstanten werden wie folgt bestimmt:

wobei R0 = 8,314472 J/(mol K), die molare Gaskonstante nach der letztgültigen Fassung von Codata-98 und Md = 18,015257 g/mol, die Molare Masse des reinen Wassers. Damit erhält man

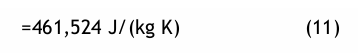
Für die reine trockene atmosphärische Luft ist Ml = 28,963 g/mol und damit

Das Verhältnis der speziellen Gaskonstanten verhält sich reziprok dem Verhältnis der Molmassen und ist
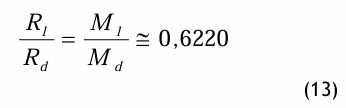
Sättigungsdampfdruck
Im Sättigungszustand ist φ = 1 und die Sättigungstemperatur erreicht den Taupunkt: T = Tτ. Über den Taupunkt hinaus wird überschüssiger Wasserdampf z.B. in Form von Tröpfchen kondensieren. Dieser Taupunkt kann z.B. mit sog. Taupunktspiegeln gemessen werden. Der Wassergehalt im Sättigungszustand, xs, beträgt dann
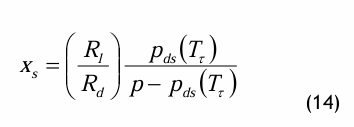
In der Thermodynamik wird üblicherweise der Wassergehalt benutzt. Darauf basiert auch das sog. Mollier-Diagramm der feuchten Luft, auch als h-x-Diagramm bekannt.
Sättigungsdampfdruck
Der Sättigungsdampfdruck pds (T) wird definiert als der Druck des reinen Dampfes, der ohne jegliche Fremdgasbeimischung im Gleichgewicht mit der ebenen Oberfläche seiner eigenen, reinen Flüssigkeit bzw. Eises steht. Der so definierte Sättigungsdampfdruck des reinen Wassers, auch Sattdampf druck oder nur Dampfdruck genannt, hängt allein von der Temperatur ab.
Die Dampfdruckkurve des Wassers, die sog. p-T Kurve, ist in den vergangenen 150 Jahren dem wachsenden Erkenntnisstand entsprechend immer wieder neu formuliert worden. Als Referenzgleichung gilt heute die Formulierung aus dem Jahre 1997 der International Association for the Properties of Water and Steam, kurz IAPWS1 genannt. Diese Gleichung kann zwar ohne große Schwierigkeiten z.B. in Excel programmiert werden, soll aber aus Gründen einer erforderlichen längeren Erläuterung hier nicht dargestellt werden. Stattdessen soll die einfache Magnus-Gleichung aus der DIN 50 010-2 benutzt werden, die zwar den hohen Anforderung der IAPWS nicht entspricht, aber hinreichend genaue Werte liefert und auch von der ISO 8573-3 zitiert bzw. benutzt wird.
Die Magnus-Gleichung ist eine einfach aufgebaute Dampfdruckgleichung, die sich dadurch auszeichnet, dass sie explizit nach der Temperatur umgestellt werden kann: die sog. Umkehrgleichung. Die Magnus-Gleichung lautet wie folgt:
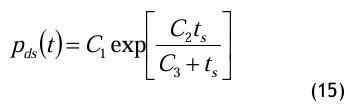
und die entsprechende Umkehrgleichung
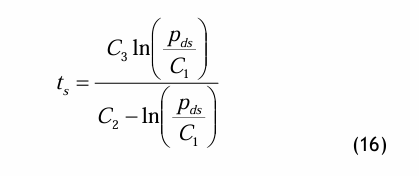
Die Konstanten sind in der folgenden Tabelle erläutert.
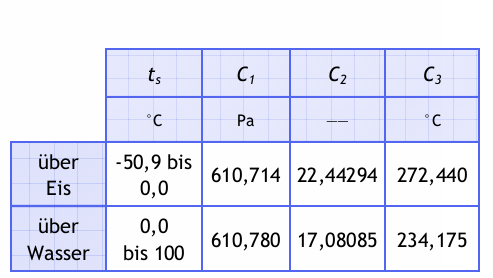
Tabelle 1 Konstanten der Magnus-Gleichung
Massenkonzentration
Volumenkonzentration
In der Ph. Eur. 4 wird der Grenzwert des Wasser dampfgehaltes als Volumenkonzentration CV an gegeben. Die Volumenkonzentration, auch Volumenanteil oder Raumanteil genannt, für den Wasserdampf wird wie folgt definiert:
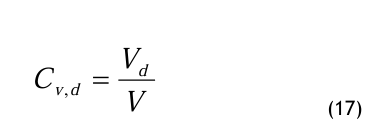
Der Volumenanteil ist also eine dimensionslose Größe, die das Partialvolumen Vd des Wasserdampfes ins Verhältnis zum Gesamtvolumen V setzt. Üblicherweise gibt man den Volumenanteil in Prozent (%) oder Promille (0/00) an. Bei sehr kleinen Werten benutzt man
- parts per million = ppmv = 1/1.000.000 = 10−6
- parts per billion = ppbv = 1/1.000.000.000 = 10−9
Der Volumenanteil bei Druckgasfeuchte muss für die Messung in eine einfachere Form überführt werden. Dazu dient abermals das Daltonsche Gesetz:

Daraus erhält man in einfacher Weise die folgen de Gleichung:
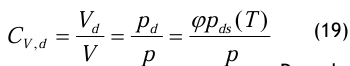
Die Druckgasfeuchte wird immer auf den Taupunkt bezogen und damit erhält man:
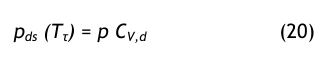
Aus der Umkehrgleichung der Dampfdruckformel erhält man den Drucktaupunkt:
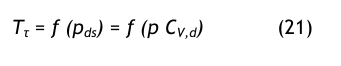
Massenkonzentration
Die Massenkonzentration oder auch der Massenanteil Cm,d wird wie folgt definiert:
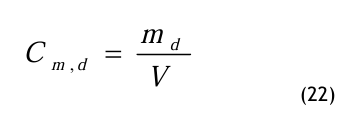
Der Massenanteil ist eine dimensionsbehaftete Größe in kg/m3, auch g/m3, mg/m3 bzw. μg/m3. Die Masse des Dampfes md wird ins Verhältnis gesetzt zum Gesamtvolumen V des feuchten Druckgases. Zur Umrechnung können der Partial dampfdruck oder der Volumenanteil dienen.
Bezogen auf den Partialdampfdruck
Die Umrechnung erfolgt mit Hilfe des Daltonschen Gesetzes in der Form der Gl.(3):
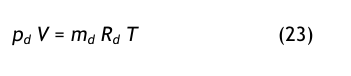
Daraus folgt
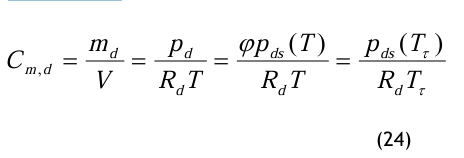
Die Berechnung des Massenanteiles erfolgt über den Drucktaupunkt bzw. den damit errechneten Partialdampfdruck.
Bezogen auf Druck und Volumenanteil
Die Umrechnung erfolgt auch hier mit Hilfe des Daltonschen Gesetzes in der folgenden Form:
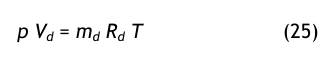
Daraus erhält man zunächst
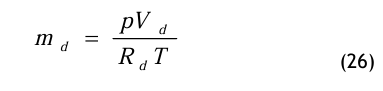
und eingesetzt in Gl.(22) erhält man damit
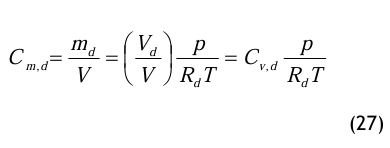
Beispielrechnung
Zur Erläuterung der beschriebenen Umrechnungsgleichungen soll ein Beispiel vollständig durchgearbeitet werden. Gemäß Ph. Eur. 4 (Aermedicalis, pp. 591- 594) wird der Grenzwert für die Feuchte in der pharmazeutischen Druckluft bzw. auch im Stickstoff wie folgt angegeben:

Leider hat die Ph. Eur. 4 nicht gesagt bei welchem Druck dieser Wert nachzuweisen ist. Es wird deshalb ein Druck von 1 atm = 101325 Pa angenommen.
Diese Festlegung erscheint auch plausibel, da nach 2.1.6, pp. 19 der Ph. Eur. 4 der Nachweis mit Hilfe von Water Vapour Detector Tubes durchgeführt werden soll und dieser Nachweis üblicherweise bei atmosphärischen Druckbedingungen geschieht, siehe auch Fig.(2.1.6-1) der Ph. Eur. 4. Die Temperatur wird mit t = 20◦C oder T = 293,15 K angenommen.
Umrechnung von Volumenanteil in Massenanteil
Bei einem Druck von p = 101 325 Pa erhält man gemäß Gl.(27)

Der Massenanteil beträgt also Cm,d = 50,2 mg/m3. Diese Umrechnung ist deshalb erforderlich, um das Prüfröhrchen entsprechend seinem Messbereich im mg/m3 auszuwählen.
Umrechnung auf den Drucktaupunkt
Der angegebene Volumenanteil der Feuchte von 67 ppmv wird umgerechnet in den Drucktaupunkt. Zunächst muss gemäß der Gl.(20) der Partialdampfdruck beim Drucktaupunkt berechnet werden. Dann kann gemäß der Umkehrgleichung (16) der Magnus-Gleichung die Sättigungstemperatur berechnet werden, die gleichbedeutend mit der Drucktaupunkttemperatur ist. Die ses Verfahren hängt natürlich vom Referenz druck ab.
Referenzdruck p = 1 atm
Gemäß der Gl.(20) ist
pds = 101 325 * (67 × 10−6) ≅ 6, 7888 Pa (30) Damit geht man in die Gl.(16), wobei die Magnus-Konstanten für den Fall über Eis gewählt werden und folgendes Zwischenergebnis benutzt wird:
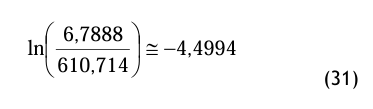
und erhält damit
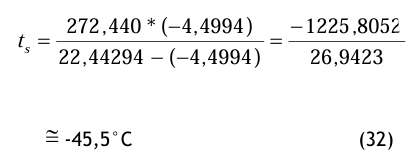
Referenzdruck p = 8 bar
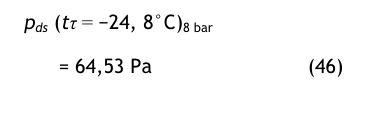
Die gleiche Berechnung für 8 bar ergibt folgende Werte:
pds = 800 000 * (67 × 10−6) ≅ 53, 6 Pa (33)
mit dem Zwischenergebnis
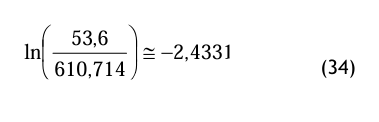
und damit
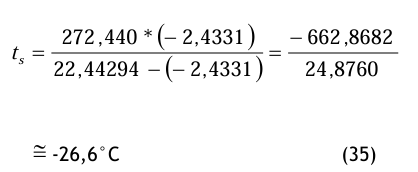
Diesen Drucktaupunkt würde man mit einem entsprechenden Messgerät in der Druckluftleitung bei einem Druck von 8 bar (absolut) messen.
Umrechnung auf Sättigungswassergehalt
Referenzdruck p = 1 atm
Der Sättigungswassergehalt wird gemäß Gl.(14) berechnet:

Referenzdruck p = 8 bar
Die gleiche Berechnung für 8 bar ergibt den folgenden Wert:

In beiden Fällen bleibt der Wassergehalt bei Druckänderungen konstant und beträgt
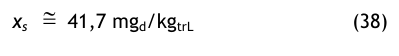
Unsicherheit der Messung mit Drägerröhrchen
In der Ph. Eur. 4 wird die Messung der Druckgasfeuchte von 67 ppmv mit Hilfe von Drägerröhrchen empfohlen. Das entsprechende Dräger Prüfröhrchen weist die folgenden Merkmale auf:
- Prüfröhrchen-Kennzeichen: Wasserdampf 5/a-P
- Standardmessbereich: 5 bis 200 mg/m3
- Prüfvolumen: 50 L
- Probenabmestrom: 2 L/min
- Dauer der Messung: 25 min
- Standardabweichung: ± 15…20 %
- Farbumschlag: gelb → rotbraun
Im unteren Messbereich (67 ppmv = 50,2 mg/m3) sollte man eher mit einer Messunsicherheit von ± 20 % rechnen. Die gesamte Unsicherheit muss auch die Messunsicherheit der Volumenstrommessung in das Budget einbeziehen: geschätzt ± 20 %, wobei die Unsicherheit der Zeitmessung vernachlässigt wurde.
Die geschätzte Gesamtunsicherheit beträgt so mit:

Die gesamte Unsicherheit in der Einheit ppmv beträgt dann

Die gesamte Unsicherheit in der Einheit mg/m3 bei atmosphärischem Druck beträgt entsprechend

Der untere und obere Wert des Feuchtegehaltes wird nun in Drucktaupunkt beim Druck von 1 atm umgerechnet nach dem oben gezeigten Rechenweg:

und

Die beiden Drucktaupunkte weichen um ein Δt = 4,8 K voneinander ab.
Drucktaupunkt-MessgerätTesto®
Ausgehend von einem Drucktaupunkt-Messgerät der Fa. Testo würde man bei einem Feuchtegrenzwert von 67 ppmv den folgenden Drucktaupunkt bei 8 bar messen:
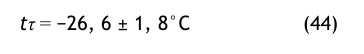
Bei dem unteren und oberen Drucktaupunkt wird bezogen auf p = 8 bar der Sättigungsdampfdruck nach der Magnus-Gleichung berechnet
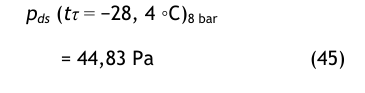
und
Diese Werte werden auf den Druck p = 101 325 Pa umgerechnet mit den folgenden Ergebnissen:

und

Die Sättigungswerte bei atmosphärischem Druck werden dann umgerechnet in Volumenanteile:
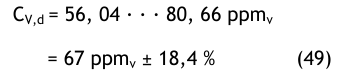
Man kann erkennen, dass die Volumenanteile mit der Methode Drucktaupunktbestimmung genauer gemessen werden können als mit der Methode Prüfröhrchen.
Trotzdem besitzt die erste Methode eine immer noch erhebliche Messunsicherheit, die bei der Messung der sehr geringen relativen Feuchte von ca. 2% hervorgerufen wird. Der große Vorteil der Drucktaupunktbestimmung liegt darin, dass der gesamte Messbereich von −55°C bis +20°C kontinuierlich durch gemessen werden kann. Bei der Prüfröhrchen-Methode müssen unter Umständen 4 verschiedene Prüfröhrchen benutzt werden, um diesen Messbereich zu überdecken.
Literatur
1
Alle IAPWS-Formulierungen über die stofflichen Eigenschaften des Wassers und des Dampfes stehen im Internet frei zur Verfügung unter den so genannten Releases der IAPWS: www.iapws.org
Validierung und Versuchsplanung
Die Biotechnologie ist einer der innovativsten Industrie zweige in Deutschland. Ob in der Entwicklung neuer Wirkstoffe oder der Herstellung von so genannten Validierungschargen für die Zulassung neuer Arzneimittel, täglich werden viele Versuche durchgeführt. Ein effizienter Einsatz von Zeit, verfügbaren Rohstoffen und Personal ist unabdingbar für den Erfolg des Unternehmens. Über den Aufwand für die Validierung herrschen unterschiedliche Auffassungen, welche hier angesprochen werden.
Wie viele Validierungschargen sind notwendig?
Die Meinung, dass man genau drei Validierungschargen herstellen müsste, ist weitverbreitet. Wo her stammt diese Zahl? Man findet bei der EMEA (1) den Hinweis, dass für nicht standardisierte Verfahren, insbesondere wenn Sterilisationsschritte oder aseptische Arbeitsweisen angewendet werden, mindestens drei aufeinander folgende Chargen im Produktionsmaßstab herzustellen sind. Für andere nicht standardisierte Verfahren können ein bis zwei Chargen im Produktionsmaß stab ausreichen, wenn aus der vorhergehenden Pilotphase aus reichend Daten vorhanden sind.
Die FDA hat in ihrem „Validation Guide“ (2) grundsätzlich drei Chargen für die Validierung gefordert. Und auch im EG-Leitfaden der Guten Herstellungspraxis wird im zutreffenden Anhang 15, „Zulassungsprüfung und Validierung“ (3), eine Anzahl von drei aufeinander folgenden Chargen als
„allgemein zulässig“ beschrieben.
Bei den Validierungschargen wird immer davon ausgegangen, dass diese unter routinemäßigen Bedingungen hergestellt werden. Gleichzeitig wird jedoch in den genannten Texten immer darauf verwiesen, dass die Parameter bekannt sein müssen und „die Zahl der durchgeführten Prozessläufe und die entsprechenden Beobachtungen ausreichen, um das normale Maß an Variationen und Trends feststellen zu können und um ausreichend Daten für die Bewertung zu erhalten.“(3) In den Guidelines der FDA findet man weiter den Hinweis „worstcase“ Bedingungen in die Untersuchungen einzuschließen.
An dieser Stelle tritt ein deutlicher Widerspruch zu Tage. Einerseits sollen Variationen und Trends fest gestellt werden, welche die kritischen Prozessparameter betreffen, andererseits werden 1 bis 3 Chargen gefordert.
In der Praxis wird daher häufig der Weg gegangen, dass drei Chargen hergestellt, aber keine kritischen Parameter verändert werden, auch wenn für diese in den Zulassungs unterlagen Bereiche angegeben wurden. Die Aussagekraft dürfte bei drei Untersuchungen jedoch begrenzt sein. Es ist also dem Hersteller selbst überlassen, vorab oder erst nach den Validierungschargen umfangreichere Tests zu fahren, welche die Bereiche tatsächlich abdecken und eine Aus sage über Trends erlauben.
PAT
Im September 2004 veröffentlichte die FDA eine Empfehlung, welche u.a. auch dieses Problem angeht. Unter dem Begriff Process Analytical Technologies wird ein regulatorischer Rahmen geschaffen, welcher die freiwillige Integration innovativer Technologien in die Entwicklung, Produktion und Qualitäts kontrolle von Arzneimitteln fördern soll (4). Hier wird u.a. auch auf die Strategie zur Durchführung von Versuchen eingegangen und die Anwendung der Versuchsplanung (DOE – Design of Experiments) empfohlen. Die Richtlinie über die Validierung befindet sich daher zurzeit in Revision. Eine ausführliche Empfehlung existiert daher noch nicht.
Jedoch sozusagen als Reaktion auf die PAT-Initiative wurde im CFR 490.100 (5) der Begriff Konformitätschargen (confomance batches) geprägt:
„Before commercial distribution begins, a manufacturer is expected to have accumulated enough data and knowledge about the commer cial production process to support post-approval product distribution. Normally, this is achieved after satisfactory product and process development, scale-up studies, equipment and system qualification, and the successful completion of the initial conformance batches. Conformance batches (sometimes referred to as „validation“ batches and demonstration batches) are prepared to demonstrate that, under normal conditions and defined ranges of operating parameters, the commercial scale process appears to make acceptable product. Prior to the manufacture of the conformance batches the manufacturer should have identified and controlled all critical sources of variability.“
Für Arzneimittelhersteller, die für den US-Markt produzieren wollen, entfällt damit die spezifische Forderung nach drei Konformitätschargen. Nun müssen die vorhandenen Untersuchungen so für die Beweisführung eingesetzt werden, das die kritischen Parameter und alle Ursachen ihrer Variabilität erkannt wurden und beherrscht werden. Damit sind die Entwicklung und die Produktion wieder näher zusammengerückt, denn Validierung kann nur noch als abschließender Teil der Produktentwicklung angesehen werden. Eine der Konsequenzen ist, dass die Durchführung und Planung in ein Gesamtkonzept eingeordnet wird, welches auch eine statistisch begründete Versuchsplanung enthält.
Versuchsplanung
Zum Verständnis der Vorgehensweise für die Validierung soll das Prinzip der Versuchsplanung kurz vor gestellt werden. In jedem Prozess gibt es wichtige und weniger wichtige Einflussfaktoren. Betrachtet man beispielsweise einen Fermentationsprozess, so werden beispielsweise folgende Parameter einen Einfluss ausüben:
- Temperatur
- pH-Wert im Nährmedium
- Zusammensetzung des Nährmediums
- Verhältnis Inoculum/Medium
- Durchmischung (z.B. Rührer drehzahl)
- Sauerstoffzufuhr
Je nachdem, ob die Einflussgrößen variiert werden können oder nicht, spricht man von Steuergrößen bzw. Störgrößen. Letztere müssen betrachtet werden, wenn robuste Prozesse entwickelt werden.
Für jeden Einflussfaktor ergibt sich ein Gültigkeitsbereich innerhalb erlaubter Grenzen. Berücksichtigt man im Versuch nur bestimmte Werte innerhalb dieser Grenzen, spricht man von Faktorstufen.
Nehmen wir an, dass nur die Temperatur und der pH-Wert als kritische Parameter identifiziert wurden. Wie könnten beide Faktoren in einer Validierung eingestellt werden? Um den Bereich abzudecken, entscheiden wir uns für die obere und untere Bereichsgrenze. Berücksichtigt man jetzt alle möglichen Kombinationen, so erhält man vier Versuche.

Zunächst soll demonstriert werden, wie der entsprechende Plan kon struiert wird. Dazu wird in einer Tabelle für jeden Faktor eine Spal te angelegt. Tabelle 1 enthält zu erst so viele Zeilen wie Versuche notwendig sind.
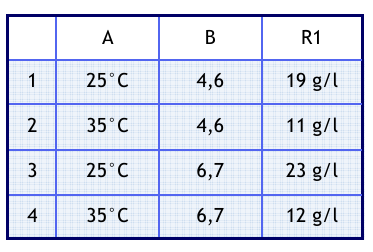
Tabelle 1: Beispiel für einen vollständigen Faktorenplan
Weiter nehmen wir folgende Werte an: Temperatur (Faktor A) im Bereich von 25°C bis 35°C; pH-Wert (Faktor B) im Bereich 4,6 bis 6,7. In der jeweiligen Zeile kann man nun die Faktorstufenkombination ablesen. In die letzte Spalte wurde außerdem ein Ergebnis (R1) für die Ausbeute eingetragen. Bei der Realisierung der Versuche ist darauf zu achten, das die Reihenfolge randomisiert wird. Zum Beispiel erhalten wir durch Auslosen die Reihenfolge: 2, 1, 3, 4
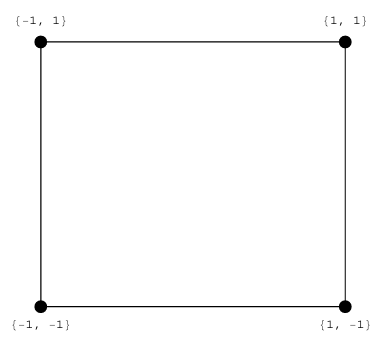
Abbildung 1: Verteilung der Versuche als Faktorenstufenkombination
Die Abbildung 1 zeigt, wie die Punkte im Versuchsraum verteilt sind. Hier wurden die Faktorstufen allgemein mit -1 für die untere und +1 für die obere kodiert. Man erkennt, dass je zwei Versuche zur Verfügung stehen, die bei einem niedrigen und einem hohen Level des betrachteten Faktors durchgeführt wurden. Will man den Effekt nun quantitativ erfassen, wird auf der unteren und der oberen Faktorstufe jeweils der Mittelwert der gemes senen Ergebnisse gebildet. An schließend bildet man die Differenz der Mittelwerte. Vergleicht man die Differenzen für die einzelnen Effekte, so erhält man eine Reihen folge. Je größer die Differenz, desto größer war auch der Einfluss des zugehörigen Faktors.
Diff A= (11+12)/2-(19+23)/2=-9
Diff B= (23+12)/2-(19+11)/2= 3
Damit folgt, dass der Einfluss des Faktors A größer ist als der des Faktors B. Weiter sieht man, dass A die Ausbeute verschlechtert, während B die Ausbeute erhöht.
Tabelle 2 zeigt die allgemeine Kodierung der Effekte und stellt gleichzeitig eine Art Rechenschema dar. Man geht so vor, dass man die kodierten Werte aus einer Spalte jeweils mit der Ergebnisspalte multipliziert, danach die Ergebnisse aufsummiert. Die Summen müssen durch die Anzahl der Werte geteilt werden, welche für die Mittelwertbildung verwendet wurden. In MS Excel lässt sich diese Berechnung schnell mit der Funktion „Summenprodukt“ ermitteln.
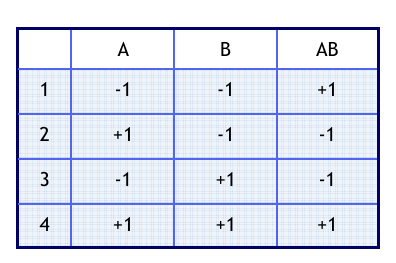
Tabelle 2: Kodierte Faktorenstufen
Die Tabelle zeigt eine weitere Spalte. Die Spalte AB ergibt sich durch die Multiplikation der Spalten A und B. Mit dem Spaltenvektor AB kann man nun die Wechselwirkungen zwischen den Faktoren nach dem gleichen Schema berechnen. Wechselwirkungen sind immer dann von Bedeutung, wenn die Veränderung eines Faktors auch einen Einfluss auf einen anderen Faktor hat. Genau dies sieht man in Abbildung 2.
Aufwandsbetrachtung
Unser Beispiel hat nur zwei Faktoren betrachtet, für welche wir einen vollständigen Faktorenplan aufgestellt haben. Will man mehr Faktoren berücksichtigen, wächst die Anzahl der Versuche exponentiell. Man kann die Anzahl der Versuche reduzieren, indem man so genannte fraktionelle Faktorenpläne aufstellt.
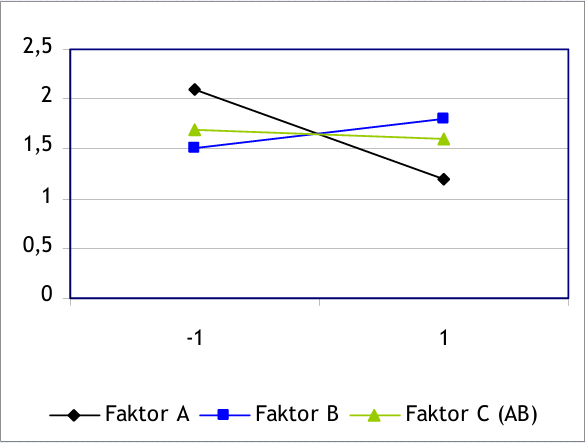
Abbildung 2: Mittelwertvergleich für die Faktoren A, B und C
Man bedient sich in diesem Falle der Wechselwirkungen. Für den Fall, dass keine Wechselwirkung zwischen A und B auftritt, kann man diese mit einem weiteren Faktor C „vermengen“. Aus der Tabelle 2 kann man in der Spalte AB ablesen, welcher Level für den Faktor C eingestellt werden muss. Auf diese Weise lassen sich 3 Faktoren mit einem Faktorenplan für zwei Faktoren untersuchen. Die Anzahl der Versuche ergibt sich wie folgt:
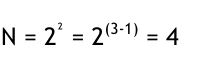
Diese Art der Reduktion ist sehr effizient, wird doch mit dem Vernachlässigen einer Wechselwirkung der gesamte Aufwand halbiert. Es ist jedoch wichtig, die Faktorenstufen exakt nach dem Muster in Ta belle 2 zu ermitteln, da nur auf diese Art eine gleichmäßige Verteilung im Versuchsraum erreicht wird. Folgendes Beispiel soll die Reduktion der Versuche anschaulich darstellen. Wir gehen von einem vollständigen Plan mit 3 Faktoren aus, so dass 8 Versuche nötig sind. Die Faktorenstufen in ihrer kodierten Form kann man durch Punkte eines Würfels darstellen.
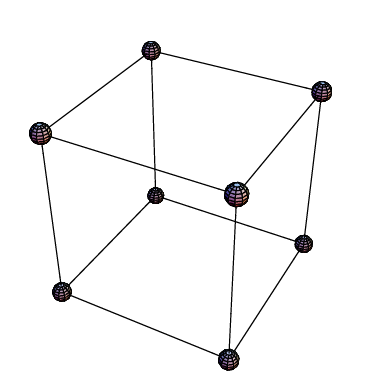
Abbildung 3: Faktorenstufenkombinationen für 3 Faktoren
Auch hier werden die Effekte dadurch ermittelt, dass Mittelwerte von allen Versuchen gebildet werden, die z.B. für Faktor A bei Level -1 und +1 durchgeführt wurden. Man kann sich das so vorstellen, dass die Ergebnisse von zwei gegenüberliegenden Würfelseiten mit einander verglichen werden. Will man nun einen reduzierten Faktorenplan aufstellen, müssen die Versuche gleichmäßig im Versuchsraum verteilt bleiben.
Abbildung 4: Auf jeder Würfelfläche befindet sich nach der Reduktion die gleiche Anzahl an Versuchen
Dadurch wird erreicht, dass die Anzahl der Versuche pro Würfelfläche auf allen Seiten auch nach der Reduktion gleich ist. Durch Projektion der Würfelecken erhält man die Darstellung in Abbildung 1. Auch in Versuchsplänen mit mehr als drei Faktoren ist diese Reduktion möglich. Es können sogar mehrere Wechselwirkungen mit neuen Faktoren vermengt werden. Zum Beispiel lässt sich mit den anfangs genannten sechs Faktoren ein 2(6-3) Plan konstruieren, der nur 8 Versuche benötigt.
Welche Aussagekraft haben die berechneten Effekte?
Die Höhe der berechneten Effekte ist ein Maß für die Höhe des Einflusses der Faktoren. Statistische Berechnungen werden oft durch so genannte Signifikanztests untermauert. Die Gültigkeit einer Aussage wird dadurch eingeschränkt, dass eine bestimmte Wahrscheinlichkeit angenommen wird, dass die Aussage falsch ist. Oft verwendet man Irrtumswahrscheinlichkeiten von 5, 1 oder 0,1 Prozent. Mit diesen berechnet man die 95, 99 und 99,9-prozentigen Vertrauensintervalle. Für die Berechnung von Vertrauensintervallen arbeitet man beispielsweise mit dem t-Test. Der t-Test ist jedoch nur anwendbar, wenn so genannte Freiheitsgrade existieren. Hinter der Berechnung der Effekte versteckt sich ein ma thematisches Modell, welches Parameter schätzt. Verwendet man das Modell für einen 2³-Plan, werden 8 Parameter geschätzt, d.h. genau so viele, wie auch Versuche durchgeführt werden. Dadurch bleibt kein Freiheitsgrad übrig.
Erst durch die Wiederholung von Versuchen werden Freiheitsgrade gewonnen. Weit verbreitet ist die vollständige Wiederholung eines Versuchsplans. Damit verdoppelt sich jedoch der Aufwand. Kennt man seinen Versuchsaufbau sehr gut und kann abschätzen, dass die Streuung zwischen Wiederholungen bei allen Faktorstufenkombinationen gleich ist, lässt sich der Aufwand reduzieren. In diesem Fall wird ein Einzelversuch zweimal wiederholt. Nun lässt sich die Streuung abschätzen und mit den gewonnen zwei Freiheitsgraden der t-Test durchführen und ein Vertrau ensintervall für die Effekte ange ben. Liegen die Effekte außerhalb dieser berechneten Vertrauensin tervalle für 95, 99 oder 99,9 % kann man die Aussage treffen, dass die Effekte mit dem jeweiligen Niveau signifikant sind. Bleiben die Effekte innerhalb der Vertrauensintervalle, sind die Effekte nicht von zufälligen Fehlern zu unterscheiden.
Für einen Gefriertrocknungsprozess sollen Konformitätschargen hergestellt werden. Es werden 2 Produkte hergestellt und in 5 verschiedenen Dosierungen von 2 bis 20 ml abgefüllt. Der Aufwand wird reduziert, indem hier nur die Abfüllung der größten und kleinsten Menge betrachtet wird. Die Gefriertrocknung erfolgt mit einem Standardprogramm, welches von einer kleineren Anlage übernommen wurde. Für die Dauer der Haupttrocknung wird durch das Scale-Up von einer möglichen Verkürzung der Zeit von 24 h auf 20 h ausgegangen. Die Größe der Chargen variiert nach Bedarf zwischen 10.000 und 20.000 Flaschen. Mit diesen Einflussgrößen wird ein fraktioneller Faktorenplan mit 2(4-1)=8 Versuchen festgelegt. Nun sind noch weitere Versuche zur Ermittlung der Versuchsstreuung nötig. Hierzu wird eine Faktorstufenkombination gewählt, von der man aus der Prozesskenntnis heraus die größte Streuung bei den unter suchten Ergebnissen erwartet. Die se Faktorstufenkombination wird mind. zweimal zusätzlich zum Versuchsplan realisiert. Der Versuchsplan enthält nun 10 Versuche. Wichtig ist, dass alle Versuche randomisiert werden, einschließlich der Zusatzversuche.
Als Zielgrößen werden der Wassergehalt, der Wirkstoffgehalt, dass Aussehen usw. gewählt. Nach Durchführung erfolgt die Berechnung der Effekte und Wechselwirkungen. Mit Hilfe der t-Statistik werden die Vertrauensgrenzen für die Effekte ermittelt. Jetzt lässt sich beurteilen, ob Effekte auftre ten. Im Falle der Validierung sollten die Effekte und Wechselwirkungen alle innerhalb der Vertrauensgrenzen liegen, also keinen signifikanten Einfluss haben. Damit wäre ein robuster Prozess nachgewiesen.

Literatur
1
EMEA, Note for Guidance on Process Validation, 2001
2
EMEA, Note for Guidance on Process Validation, 2001
3
EG-Leitfaden der Guten Herstel lungspraxis, Annex15, Qualification & Validation, 2001
4
FDA, Guideline on General Princi ples of Process Validation, 1987
5
FDA, PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing and Quality Assurance, 2004
1 Einleitung
1.1 Gegenstand
Die Inspections Group der EMEA hat am 21. September 2005 ein Arbeitspapier ver öffentlicht [8], das einige Änderungen an der z. Zt. gültigen ergänzenden Leitlinie 1, kurz Annex 1 [7], zur Diskussion stellt. Das 4-seitige Arbeitspapier wurde erst am 23.11.2005 mit einigen erläuternden Anmerkungen auf den EMEA-Server gestellt, und es wird von seiten der EMEA die Möglichkeit eingeräumt, die Änderungsvorschläge bis zum 30. April 2006 zu kommentieren. Aus diesem aktuellen Anlass möchten wir einige dieser Änderungsvorschläge diskutieren, die sich um die Begriffe Luftwechsel, Reinheitsgrade und Clean Up Period ranken.
1.2 Ziel
Das Ziel dieses Reports besteht darin, den Begriff Clean Up Period zu analysieren. Dieser Begriff steht im unmittelbaren Zusammenhang mit dem Luftwechsel und der Durchspülung von Reinräumen der Reinheitsklasse B, C und D. Nur diese Reinräume, die lüftungstechnisch mit einer turbulenten Mischströmung ausgestattet werden, unterliegen der regulatorischen Anforderung zum Nachweis der Clean Up Period. Das Verfahren zum Nachweis der Clean Up Period wird als sog. Erholzeitmessung (Recovery Test) im Abschnitt B.12 der ISO 14644-3 [12] beschrieben. Damit ist das zweite Ziel dieses Aufsatzes umrissen: die Erläuterung und Anwendung der Erholzeitmessung.
1.3 Zweck
Zeitlich gesehen wird der Nachweis der Clean Up Period im Zuge der Funktions- und Leistungsqualifizierung (OQ/PQ) kurz vor dem Beginn des Produktionsbetriebes erbracht. Über die Auslegung der RLT-Anlage wird aber bereits im Entwurfsstadium entschieden. Will man also beim Nachweis der Clean Up Period keine unangenehmen Überraschungen erleben, ist es erforderlich, bereits im Entwurfsstadium die spätere Erfüllung dieser wichtigen regulatorischen Anforderung sorgfältig einzuplanen. Der Zweck dieses Aufsatzes besteht nun darin, auf diesen wichtigen Umstand hinzuweisen.
2 Übersicht Annex1
2.1 Annex 1: Mai/September 2003
Die erste ergänzende Leitlinie für die Herstellung steriler Arzneimittel, nachfolgend kurz als Annex 1 zitiert, wurde seit ihrer Urfassung [1] aus dem Jahre 1989 (in Kraft: Januar 1992) mehrfach überarbeitet. Die letzte Überarbeitung durch die Ad Hoc GMP Inspectors Group wurde vom Pharmaceutical Committee im Mai 2003 angenommen und im September 2003 in Kraft gesetzt [7].
Sie besteht aus 20 nicht nummerierten Kapiteln, deren Überschriften durch einen größeren Schrifttypus hervorgehoben werden: PRINCIPLE, GENERAL bis QUALITY CONTROL. Die Kapitel sind meist in mehrere Abschnitte unterteilt. Diese Abschnitte sind von 1 bis 93 durchnummeriert worden und werden wahlweise section oder clause genannt, die in diesem Aufsatz zur eindeutigen Kennzeichnung mit einem Paragraphenzeichen (§) versehen sind. Gemäß der Zielsetzung betrachten wir den Abschnitt § 3 und speziell die folgenden Anmerkungen (Notes), die an der Tabelle der Reinheitsklassifikation
angefügt wurden:
- Note (b): über die kurze Freispülphase von 15-20 Minuten (Clean Up Period)
- Note (c): über die Zahl der Luftwechsel in den B-, C- und D-Bereichen
2.2 Proposals: September 2005
Die Änderungsvorschläge (proposals for amendment) kann man wie folgt zusammenfassen:
- Die Abschnitte § 3 und § 4 in General sollen ersetzt werden durch 7 neue Abschnitte: § 3 bis § 9, ebenfalls wieder unter dem Kapitel General. Diese Mehrung um 5 weitere Abschnitte entsteht vor allen Dingen dadurch, dass die bisherigen 6 Anmerkungen [Notes (a) bis (f)] zur Tabelle der Reinheitsklassifikation aufgelöst werden und nicht mehr als solche zu dieser Tabelle erscheinen, sondern als eigene Abschnitte verfasst werden.
- Die Abschnitte § 42 und § 52 in Processing sollen inhaltlich verändert werden und bilden dann die neuen Abschnitte § 47 und § 57, ebenfalls wieder unter dem Kapitel Processing.
- Der Abschnitt § 88 in Sterile Products soll inhaltlich verändert werden und bildet dann den neuen Abschnitt § 93, ebenfalls wieder unter dem Kapitel Sterile Products.
- Nach allen Änderungen entstehen dann die neu durchnummerierten Abschnitte § 1 bis § 98.
2.3 Unterteilung der reinen Bereiche
Bekanntlich muss die Produktion steriler Zubereitungen in reinen Bereichen durchgeführt werden. Diese Annex-1-Bereiche sollen von anderen Produktionsbereichen (nicht-Annex-1 Bereiche) dadurch separiert werden, dass der Personenzugang und das Einbringen von Ausrüstung und Materialien über Schleusen erfolgen soll: § 1 in Annex 1.
Nach dem Prinzip der abgetrennten Zonen werden die Annex-1-Bereiche in einen kritischen Kernbereich (A-Bereich in B-Umgebung) und in kontrollierte Nebenbereiche (C/D-Bereiche) unterteilt (§ 2: separate areas within the clean area). Es werden deshalb 4 verschiedene Reinheitsklassen unterschieden mit jeweils eigenen Anforderungen, speziell bezüglich der Lüftungskonzepte.
2.4 Unidirectional Air Flow
Die Anforderungen an die A-Bereiche werden in der Regel nur durch Laminar-Air-Flow-Systeme erfüllt. Bereits der Erfinder des Laminar-Air Flow-Prinzips, Willis WHITFIELD, hat im Jahre 1962 diese Bezeichnung selbst in die Welt gesetzt und zugleich als nicht zutreffend gekennzeichnet, weil es sich nicht um eine laminare Strömung im Sinne der Strö mungsmechanik handelt. Er hat die korrekte Bezeichnung uni-directional air flow vorgeschlagen, die dann sehr viel später im FED STD-209-E und in den ISO-Standard 14644 übernommen wurde.
Der Begriff laminar air flow ist aber so tief in der Fachsprache der Reinraumtechnik verankert, dass auch im Annex 1 beide Bezeichnungen einträchtig nebeneinander verwendet werden (siehe § 3, Erläuterungen zu Grade A). Im Deutschen wird der Begriff turbulenzarme Verdrängungsströmung (kurz TAV genannt) verwendet, der aus strömungstechnischer Sicht sogar eine genauere Bezeichnung für das amerikanische uni-directional air flow ist, siehe VDI 2083-2 [13].
2.5 Non-Unidirectional Air Flow
Das zweite Grundkonzept der Reinraumtechnik ist das ”non-unidirectional air flow”-System, bei uns turbulente Mischströmung (kurz: TMS) genannt. Dieses System ist dadurch gekennzeichnet, dass die Luft aus einem oder mehreren, in der Regel mit HEPA-Filtern bestückten Zuluft-Auslässen in den Raum einströmt (Erstluft, first air) und die vorhandene Kontamination verdünnt.
Die Anforderungen an die B-, C- und D-Bereiche bestehen in der Einhaltung einer bestimmten Luftwechselhäufigkeit bzw. einer bestimmten Clean Up Period. Im EG-Leitfaden wird zunächst stillschweigend vorausgesetzt, dass diese Bereiche mit einem turbulenten Misch- oder Verdünnungssystem (TMS) ausgestattet werden. In den ersten 4 Fassungen des EG-Leitfadens wurden mehr als 20 Luftwechsel pro Stunde gefordert. Diese Anforderung wurde häufig kritisiert, weil dies hohe Investitions- und Betriebskosten für die pharmazeutischen Unternehmen bedeutete [16].
Seit der Überarbeitung des Annex 1 im Jahre 1997 wurde die Festlegung auf einen 20-fachen Luftwechsel aufgegeben und durch eine verklausulierte Empfehlung ersetzt, die eine Auslegung des Luftwechsels in Abhängigkeit von der Raumgröße, Raumausstattung und Anzahl der Personen erforderlich macht. Hinzu kam eine Präzisierung des Begriffes ”Short Clean Up Period”, die auf einen Richtwert von 15 bis 20 Minuten festgelegt wurde.
3 Regulatorische Anforderungen
3.1 Annex 1 (September 2003), § 3, Note (b)
3.1.1 Der englische Originaltext
The particulate conditions given in the table for the ”at rest” state should be achieved after a short ”clean up” period of 15-20 minutes (guidance value) in an unmanned state after completion of operations.
3.1.2 Übersetzung
Die Grenzwerte für die Partikelanzahl konzentration, die in der Tabelle für den Ruhezustand festgelegt sind, müssen nach einer kurzen Freispülphase von 15-20 Minuten (Richtwert) wieder erreicht werden, und zwar ohne Anwesenheit von Personen und nach Abschluss aller Betriebstätigkeiten.
3.2 Proposals, September 2005, § 7
3.2.1 Der englische Originaltext
The particle limits given in the table for the ”at rest” state should be achieved after a short ”clean up” period of 15-20 minutes (guidance value) in an unmanned state after completion of operations.
3.2.2 Anmerkungen
Die Note (b) soll ersetzt werden durch einen eigenen Abschnitt § 7. Der Inhalt bezüglich der Clean Up Period ist geblieben bis auf eine Präzisierung: aus den particulate conditions sollen particle limits werden.
3.3 Annex 1 (September 2003), § 3, Note (c)
3.3.1 Der englische Originaltext
Der erste Satzteil: In order to reach the B, C and D air grades, the number of air changes should be related to the size of the room and the equipment and personnel present in the room.
Der zweite Satzteil: The air system should be provided with appropriate terminal filters such as HEPA for grades A, B and C.
3.3.2 Übersetzung
Der erste Satzteil: Um die Luftreinheit in den Räumen der Reinheitsklassen B, C und D zu erreichen, muss die Anzahl der Luftwechsel in Beziehung gesetzt werden zur Größe des Raumes und zur Ausrüstung, die im Raum vorhanden ist bzw. zur Anzahl der Personen, die im Raum anwesend sind.
Der zweite Satzteil: Das Lüftungssystem in den Reinräumen der Reinheitsklasse A, B und C sollte mit geeigneten endständigen Filtern wie z.B. HEPA-Filtern ausgerüstet sein.
3.4 Proposals, September 2005
In den Änderungsvorschlägen soll der Passus über die Anzahl der Luftwechsel und die Ausstattung mit HEPA-Filtern gestrichen werden.
3.5 Bemerkungen
In der ersten Fassung des Annex 1 wurde die Ausstattung mit HEPA-Filtern für die Reinheitsbereiche A, B, C und D und ein mehr als 20-facher Luftwechsel für die Reinheitsbereiche B, C und D festgeschrieben. Zumindest in dieser Hinsicht wurde eine vollständige Übereinstimmung mit der Aseptic Guide der FDA erzielt. Diese Anforderungen gelten für die FDA noch heute, siehe Abschnitt IV Buildings and Facilities in [9], während sie im Annex 1 mit jeder Änderung nach und nach weitergefasst bzw. verallgemeinert wurden, und gemäß den Proposals soll es demnächst überhaupt keine Anforderung bezüglich Luftwechselhäufigkeit und Ausstattung mit HEPA-Filtern mehr geben.
Nach dem Stand und den Regeln der Technik kann man auf eine ausdrücklich gegebene Anforderung nach dem Einsatz von HEPA-Filtern in den Reinräumen natürlich verzichten, da sonst die Erfüllung des Reinheitsgrades nicht möglich wäre. Ebenso kann man auf die Festlegung einer bestimmten Luftwechselhäufigkeit in den Reinräumen verzichten, wenn eine bestimmte Clean Up Period vorgeschrieben wird. Dies wird in den nächsten Abschnitten gezeigt.
4 Luftwechsel und Erholzeit
4.1 Max von PETTENKOFER
Der Begriff Luftwechsel wurde von Max von PETTENKOFER1 in einem Aufsatz [15] aus dem Jahre 1886 erstmals geprägt. Er stellte die Hygieneforderung auf, dass Wohnräume gelüftet werden sollen. Die komplette Raumluft sollte einmal stündlich gegen Frischluft ausgetauscht werden. Dadurch sollten die Ausdünstungen der Menschen (und die der Nachttöpfe!) aus der Wohnung ausgetrieben werden, ebenso die durch Holzfeuerung, Kochen und Waschen ver ursachten Abgase, Dämpfe und Gerüche. PETTENKOFER, der eigentlich Chemiker war, hat gezeigt, dass der CO2-Ausstoß durch den Menschen selbst und durch seine Art der Energieerzeugung in ungelüfteten Räumen schädlich war und hat den CO2-Gehalt der Raumluft zum Maßstab der Lüftungshäufigkeit erhoben. Dieser CO2-Maßstab ist in die VDI Lüftungsregeln eingegangen, welche später als DIN 1946 noch bis in die 1980-er Jahre gültig waren.
4.2 Begriffe
Der Begriff des Luftwechsels hat sich im Laufe der Jahre gewandelt. Aus dem Frischluftwechsel wurde der Außenluftwechsel, weil der Begriff Frischluft eine Luftqualität suggeriert, die die Außenluft (z.B. in Städten) nicht besitzt.
Zur Präzisierung wurde der Begriff Raumluftwechsel eingeführt, da der Luftaustausch sich ja auf den Luftinhalt des Raumes bezieht. In den modernen RLT-Anlagen wird der Raumluftwechsel auch nicht mehr durch reine Außenluft bewirkt, sondern durch ein Gemisch von Außenluft und rezirkulierter Raumluft (Umluft genannt). Dieses Gemisch wird dann Zuluft genannt, so dass wir heute von einem Zuluft-Raumluftwechsel sprechen. Diese Zuluft kann in besonderen Fällen aus reiner Außenluft bestehen. Wir bezeichnen dies mit dem Begriff Außenluft-Raumluftwechsel. In der Regel sprechen wir aber von einer Zuluft, die nur einen geringen Anteil von Außenluft (make-up air) enthält, gerade soviel, dass die unvermeidlichen Raumleckagen, die Prozessfortluftmengen und bestimmte hygienische Anforderungen (Außenluftrate) abgedeckt sind.
4.3 Zuluft-Raumluftwechsel
Die heute in den Regelwerken benutzten Begriffe wie Häufigkeit des Luftwechsels (EG Leitfaden) oder Air Changes per Hour (Aseptic Guide der FDA) sollten als stündlicher Zuluft Raumluftwechsel verstanden werden:
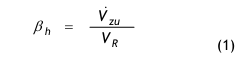
wobei Vzu den Zuluft-Volumenstrom in m³/h darstellt, der einem Raum aktiv zugeführt wird und VR das Volumen des leeren Raumes in m³. Der stündliche Zuluft-Raumluftwechsel βh ist damit eine volumenspezifische Flussgröße mit der Einheit 1/h.
In der Regel werden die Reinräume mehrere Zuluft-Auslässe aufweisen. Der dem Raum insgesamt zugeführte Volumenstrom besteht aus der Summe der Volumenströme aller Zuluft Auslässe:
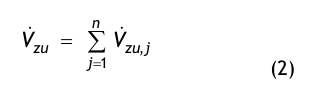
wobei n = Anzahl der Zuluft-Auslässe ist.
Unter dem Zusatz “aktiv zugeführt“ soll die direkte Raumeinspeisung der Zuluft aus einer RLT-Anlage verstanden werden. Leckluftströme, die beispielsweise über Türspalte dem betrachteten Raum aufgrund von Raumdruckdifferenzen aus benachbarten Räumen zufließen können, sollten nicht dem Zuluft-Raum luftwechsel zugerechnet werden. Diese Leckluftströme könnten kontaminiert sein und würden so nicht zu einer Verdünnung beitragen.
4.4 Lüftungseffektivität
Der Zuluft-Raumluftwechsel ist ein erstes Maß für die Raumdurchspülung. Die Zuluft besitzt aufgrund ihrer Geschwindigkeit, mit der sie aus den Auslässen in den Raum eintritt, einen Impuls, der groß genug ist, um eine Raumströmung in Gang zu setzen und aufrecht zu erhalten. Auf diese Weise mischt sich die durch die HEPA-Filter hoch gereinigte Luft, auch Erstluft oder first air genannt, mit der durch den Prozess und die Personen kontaminierten Raumluft und bewirkt somit eine Verdünnung der Kontamination. Dieses Lüftungsprinzip wird, wie oben bereits beschrieben, turbulente Mischströmung genannt.
Der Spülgrad oder die Lüftungseffektivität ist ein weiteres Maß dafür, wie gut die Raumluft mit der Erstluft gemischt und damit die Kontamination verdünnt wird. Die Lüftungseffektivität, engl.: ventilation efficiency oder ventilation effectiveness, wird durch die folgende Gleichung definiert:
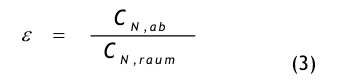
wobei CN,ab die mittlere Partikelanzahlkonzentration in der Abluft bedeutet und CN,raum die mittlere Partikelanzahlkonzentration in der Raumluft. Bei idealer Durchmischung ist ε= 1 und die Qualität der Raumluft und die der Abluft sind gleich. Bei nicht idealer Durchmischung ist CN,raum > CN,ab und ε < 1.
Man fragt sich, wie sich überhaupt Qualitätsunterschiede in der Raumluft und Abluft ausbilden können, da es doch die „gleiche“ Luft ist. Im Raum bilden sich Stagnationszonen aus, d. h. großvolumige ortsfeste Drehströmungen der Raumluft, in denen sich die Kontamination anreichern kann, siehe Abb.(1).
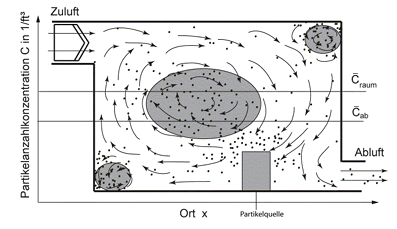
Abb.1: Schematische Darstellung des Spülgrades
Es entstehen örtliche Konzentrationsdifferenzen im Raum, weil die Erstluft nicht in die Stagnationszonen hineingelangt. Sobald aber die Raumluft in den Abluftkanal einströmt, wird sie aufgrund der turbulenten Kanalströmung intensiv durchmischt. Dadurch, dass nun die Anteile der „unverbrauchten“ Erstluft mit der kontaminierten Raumluft intensiv gemischt werden, sinkt die Partikelanzahlkonzentration im Abluftkanal gegenüber der, die im Raum im Volumenmittel vorherrscht. Dieser Spüleffekt wurde Ende der 1940-er Jahre entdeckt [14] und wird bis heute im Rahmen der Indoor Air Quality weiter untersucht.
Wie oben gesagt: nur die Zuluft kann diesen Verdünnungseffekt bewirken. Die Abluft übt dagegen nur einen geringen Einfluss auf die Raumströmung aus. Das hängt damit zusammen, dass die Ansauggeschwindigkeit bereits in sehr kurzem Abstand vor den Abluft-Einlässen auf Null absinkt. Bildlich gesprochen: man kann eine Kerze auspusten, aber nicht ”aus”-saugen. Die Abluft wirkt in diesem Sinne nur passiv auf die Raumströmung ein. Durch eine geschickte Anordnung der Abluft-Einlässe können Stagnationszonen im Raum leer gesaugt werden, d.h. die Abluft wirkt gewissermaßen als eine Grenzschicht-Absaugung.
5 Recoveryfunktion
5.1 Bilanzgleichung
Aus einer raumseitigen Partikelstrombilanz, d.h. einer Bilanz aller dem Raum zu- und abströmenden Aerosolteilchen, gewinnt man die Recoveryfunktion, auch Abklingfunktion oder Erholzeitgleichung genannt:
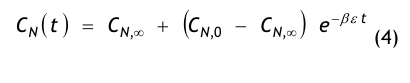
Die in der Modellgleichung verwendeten Größen haben die folgende Bedeutung:
- CN(t) = die Partikelkonzentration, die zur Zeit t im Raum vorhanden ist,
- CN,0 = die Anfangskonzentration, d.h. die im Raum vorhandene Partikelkonzentration zur Zeit t = 0,
- CN,∞= die Endkonzentration, d.h. die im Raum vorhandene Partikelkonzentration nach sehr langer Spülzeit (t → ∞), der sog. stationäre Endwert oder Ruhewert,
- t = die Zeit in Minuten (min),
- β = der Luftwechsel in 1/min, der aus dem stündlichen Luftwechsel βh abgeleitet wird:
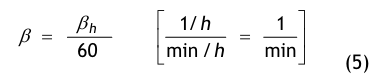
- ε = eine dimensionslose Größe, die den Spülgrad oder die Lüftungseffektivität bzw. ventilation effectiveness gemäß den o.g. Erklärungen zu Gl.(3) beschreibt.
Die Größe CN steht hier allgemein für die mittlere Partikelanzahlkonzentration im Raum, wobei wir unter dem Mittelwert das Volumenmittel verstehen. Unter dem Begriff Partikelanzahlkonzentration, manchmal auch Anzahldichte genannt, versteht man die Anzahl der Aerosolteilchen, die in einem Kon trollvolumen dispergiert sind. Da die Anzahl keine Dimension besitzt, ist die Einheit der Anzahlkonzentration 1/m³ bzw. 1/ft³. Noch genauer sollten wir von einer kumulativen Partikelanzahlkonzentration (Häufigkeitssumme) sprechen, weil wir alle Aerosolteilchen be trachten, deren Teilchengröße Dp gleich oder
größer als eine betrachtete Teilchengröße D*p ist. Häufig wird als betrachtete Partikelgröße D*p = 0,5 m gewählt, so dass wir von einer kumulativen Partikelanzahlkonzentration mit Dp 0,5 µm sprechen.
5.2 Darstellung
In Abb.(2) ist die Recoveryfunktion beispielhaft für einen B-Raum in einem halblogarithmischen Netz dargestellt. Auf der y-Achse wird die Partikelanzahlkonzentration CN(t) in 1/m³ aufgetragen. Weil der Wertebereich der Anzahlkonzentration mehrere Dekaden über spannen kann, wird die y-Achse logarithmisch geteilt. Auf der x-Achse wird die Zeit t in Minuten aufgetragen. Diese Achse wird linear geteilt.
Der Kurvenverlauf der Recoveryfunktion beginnt bei der Zeit t = 0 mit dem Anfangswert CN,0; im Beispiel wurde CN,0 = 350.000 /m³ gewählt. Die Kurve fällt zunächst steil ab und geht dann mit gleichmäßiger Krümmung in den stationären Wert CN, über. Nach sehr langer, theoretisch unendlich langer Zeit t→ ∞ wird der stationäre Endwert CN∞, = 1.750 1/m³ erreicht.
Nach der Definition der Clean Up Period ist der Wert CN,0 als der Grenzwert der Partikelanzahlkonzentration im Raum für den Betriebszustand in Operation zu betrachten:
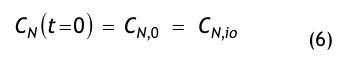
(Nach etwa 20 min ist der Wert der Partikelkonzentration auf CN,ar = 3.500 1/m³ abgesunken. Die Größe CN,ar bezeichnen wir als die mittlere Partikelkonzentration im Raum im Betriebzustand at rest. Der Verdünnungsfaktor soll mit φ bezeichnet und wie folgt definiert werden:

Im gewählten speziellen Fall beträgt der Verdünnungsfaktor:
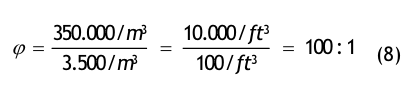
Die Zeit, die vergeht, um von der Partikelkonzentration in operation (CN,io) auf die Partikelkonzentration at rest (CN,ar) abzusinken, wird Clean Up Period genannt. Diese Größe soll allgemein mit tφ bezeichnet werden, in diesem speziellen Fall mit t0,01. Diese Bezeichnung wurde so in der ISO 14644-3 gewählt. Damit der at-rest-Zustand in dieser Zeitperiode erreicht werden kann, muss der Ruhezustand CN,∞ unterhalb des Wertes von CN,ar liegen. Diese treibende Konzentrationsdifferenz CN,ar CN,∞ soll mit Hilfe des Antriebsfaktors δ definiert werden:
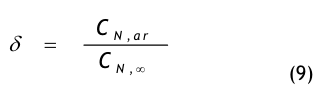
Die Gl. (4) wird wie folgt umgestellt:
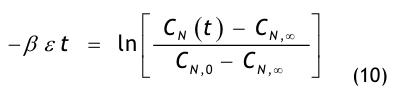
Die Konzentrationsgrößen werden mit Hilfe der Gln.(7) und (9) ersetzt:
- Die Konzentration zur Zeit t = tφ beträgt: CN(tφ ) = CN,ar.
- Die Anfangskonzentration zur Zeit t = 0 beträgt: CN,0 = CN,io = φCN,ar.
- Die Endkonzentration zur Zeit t →∞ beträgt: CN,∞ = CN,ar /δ .
Wir setzen nun diese Größen in Gl.(10) ein und erhalten ein für die Auslegung der Raumlüftung wichtiges Entwurfskriterium:
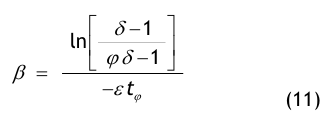
Beispiel:
Wir möchten den erforderlichen Luftwechsel β für einen B-Raum unter der Bedingung ermitteln, dass der regulatorische Wert φ = 100 für den Verdünnungsfaktor eingehalten wird und die Clean Up Period den Mindestwert von tφ 20 min erfüllt. Aus dem Maschinenlayout wurde eine Raumdurchspülung von ε = 0,75 geschätzt. Die Restkontamination wurde mit Hilfe des Filterkonzeptes, der Qualität des Außenluftaerosols und der inneren Partikel quellen auf CN,∞ ≅ 50/ft³ ( ≥ 0,5 µm) abgeschätzt, d.h. δ = 100/50 = 2. Damit ist der folgende Luftwechsel erforderlich:
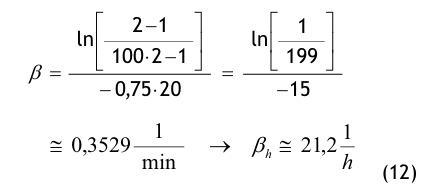
Wenn es uns gelingt, die Raumdurchspülung durch die Wahl geeigneter Zuluftauslässe so zu verbessern, dass der Spülgrad auf ε > 0,8 ansteigt, dann würde auch ein 20-facher stündlicher Luftwechsel zur Erfüllung der regulatorischen Anforderungen ausreichen, siehe Abb.(6).
5.3 Normierung
Die Recoveryfunktion kann wie folgt normiert werden:
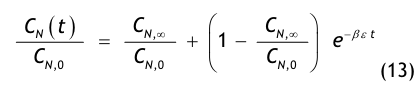
Diese Normierung bildet die Ausgangsgleichung für ein Regressionsverfahren, das zur Ermittlung der unbekannten Größen CN∞, und n = β ε aus den Werten der Erholzeitmessung dient. Bei dem Verfahren nach ISO 14644-3, Abschnitt B.12, wird CN∞, meist vernachlässigt, und es wird eine Regression nur bezüglich der Zerfallskonstante n = β ε durchgeführt nach der folgenden Gleichung:
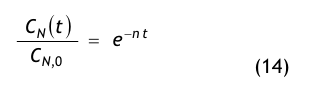
Diese Vernachlässigung (CN,∞ ≈ 0) führt in vielen Fällen zu einer Überschätzung der Clean Up Period tφ und damit zu einer Fehlinterpretation des wirklichen Freispülverhaltens des Raumes wie im nachfolgenden Beispiel noch näher ausgeführt wird.
5.4 Erläuterung
Der Ausdruck auf der linken Seite von Gl.(13) ist die dimensionslose normierte Partikelanzahlkonzentration, weil diese Größe im Anfangs zustand den Wert 1 besitzt, die sog. mathematische Norm. Auf der rechten Seite von Gl.(13) steht eine Exponentialfunktion, dessen Exponent außer der Zeit t auch das Produkt der Größen β und ε enthält. Dadurch wird unmittelbar deutlich, dass die Abnahme der mittleren Partikelanzahlkonzentration im Reinraum mit der Zeit sowohl vom Zuluft-Raumluftwechsel als auch von der Lüftungseffektivität abhängt. Eine schlechte Durchspülung des Raumes muss demnach durch einen höheren Luftwechsel kompensiert werden.
Wenn endständige HEPA-Filter benutzt werden, wird nach sehr langer Spülzeit die Partikelanzahlkonzentration auf einen Wert nahe Null zurückgehen, d.h.:
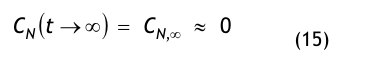
Nur dann kann auch die Abklingfunktion wie in Gl.(14) vereinfacht werden:
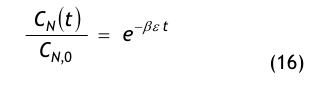
6 Beispiel
6.1 Methode
Der Nachweis der Clean Up Period wird mit Hilfe der Erholzeitmessung durchgeführt. Dabei geht man in der Regel so vor, dass zunächst die kumulative Partikelanzahlkonzentration im Raum künstlich erhöht wird. Bei laufender RLT-Anlage wird mit Hilfe eines Partikelgenerators ein Aerosolstrom für eine kurze Zeit in den Raum versprüht. Nach einer gewissen Verteilzeit ist die Partikelanzahlkonzentration im Raum soweit vergleichmäßigt und abgeklungen, dass wir den Partikelzähler einschalten und mit der Messung beginnen können.
Diese Verteilzeit ist deswegen erforderlich, um den sog. quasi-stationären Zustand zu erreichen und die Anfangskonzentration auf ein für den Partikelzähler zuträgliches Maß absinken zu lassen. Wir messen zu gewissen Zeitpunkten ti die jeweilige Konzentration CN(ti):
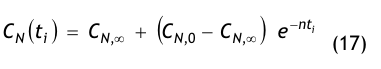
Es sollten so viele Messwerte i = 1, 2, 3, · , N gesammelt werden, dass eine geeignete statistische Auswertung durchgeführt werden kann, z.B. das GAUSSsche Regressionsverfahren der Minimierung der Abweichungsquadrate (least squares approximation). Dazu ist es sinnvoll, die Messwerte zu normieren:
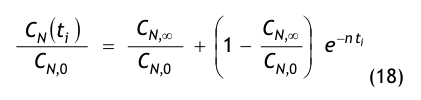
Aus der Regression können dann die gesuchten Größen CN,∞ / CN,0 und n =β ε bestimmt werden. Die Einzelheiten zum Regressionsverfahren sind im Anhang 1 dargestellt.
6.2 Ergebnis
In der Tab.(2) sind die Messwerte einer Erholzeitmessung in einer Excel-Tabelle zusammengestellt. Es wurde in einem C-Raum mit einem ca. 20-fachen stündlichen Luftwechsel gemessen mit dem Ziel, die Clean Up Period für eine 100:1-Verdünnung nachzuweisen. Der Anfangswert lag bei ca. 70.000/ft³, d.h. deutlich unterhalb des Koinzidenz-Fehlers des optischen Partikel zählers. Nach 16 Minuten wurde die Messung abgebrochen, als anhand der Werte auf dem Druckerstreifen erkennbar wurde, dass die Partikelanzahlkonzentration nicht mehr weit vom stationären Endwert lag und die Verfolgung des weiteren Absinkens der Werte keine zusätzliche Information mehr gebracht hätte.
Die so gewonnenen Messwerte wurden einer vollständigen Regression unterworfen und mit den berechneten Regressionskonstanten
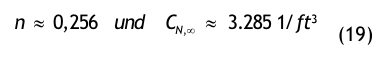
konnte die Regressionskurve gezeichnet werden, siehe Abb.(3). Man erkennt, dass sich die Messwerte sehr gut um die Regressionskurve herum gruppieren. Aus der Abb.(3) wird auch ersichtlich, dass eine 100:1-Verdünnung nicht erreicht wurde, denn dann hätten wir den Partikelzähler Konzentrationen über 300.000/ft³ aussetzen müssen. Der Nachweis der Clean Up Period muss ab hier rein rechnerisch erfolgen:
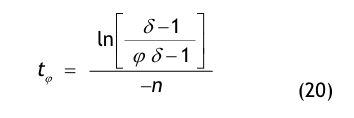
Zur Berechnung von tφ wird der Antriebsfaktor δ benötigt, der aus der Messung oder aus der Regression gewonnen wird:
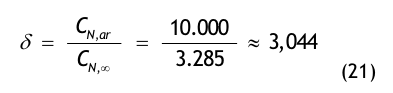
Damit erhalten wir:
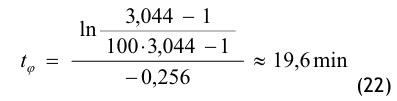
d.h. die vorgegebene Clean Up Period wird eingehalten. Eine graphische Veranschaulichung zur Auswertung nach der vollständigen Regression zeigt Abb.(5).
Nach der ISO-Methode, d.h. unter Vernach lässigung des stationären Endwertes erhalten wir mit der EXCEL-Regression n** 0,200 bzw. tφ 23 min und mit der wahren Exponential Regression n* 0,224 bzw. tφ 20,6 min. In beiden Fällen wird die vorgegebene Clean Up Period überschritten, siehe Abb.(4), man muss hinzufügen: nur scheinbar, denn das ISO Verfahren ist in den meisten Fällen nicht geeignet, die Clean Up Period aus den Messwerten einer Erholzeitmessung zu bestimmen.
7 Zusammenfassung
7.1 Regulatorischer Text
Für die Räume der Reinheitsklasse B, C und D wird nach Annex 1 die Einhaltung einer Clean Up Period von 15 … 20 min gefordert. Diese Anforderung wird in den Proposals for Amendment fast wortgleich übernommen und insofern keine Änderung der regulatorischen Absicht angestrebt.
DieClean Up Period wird allerdings dadurch zu einem prominenten Qualitätsmerkmal, dass alle weiteren Anforderungen sowohl bezüglich der Einhaltung einer bestimmten Luftwech selhäufigkeit wie auch bezüglich der Verwendung von endständigen HEPA-Filtern ersatzlos gestrichen werden sollen. Deshalb sollten die Merkmalsanforderungen besonders klar und eindeutig definiert werden. Hier setzt nun unsere Kritik an den Formulierungen in dem Annex 1 wie auch in den Proposals ein.
Aus dem kryptischen Text des § 3, Note (b) bzw. des neuen § 7 kann man herauslesen, dass sich dieClean Up Period bei den B-Räumen auf eine 100:1-Verdünnung beziehen soll, die bei den C Räumen auf eine 10:1-Verdünnung. In der Tabelle der Reinheitsklassen sind die Grenzwerte der Partikelanzahlkonzentration für die Betriebszustände at rest und in operation eindeutig benannt, so dass die Ver dünnungsfaktoren daraus abgeleitet werden können = φ CN,io / CN,ar.
Bei den D-Räumen fehlen diese Grenzwerte für den Betriebszustand in operation, d.h. wir wissen eigentlich nicht, auf welchem Verdünnungsfaktor die Clean Up Period basieren soll. Trotzdem wird im neuen § 8 die Einhaltung der empfohlenen Clean Up Period auch für D Räume gefordert.
Wir empfehlen deshalb, die Clean Up Period für die Reinheitsklasse D auf den Wert 20 min bei einem Verdünnungsfaktor φ=2:1 festzulegen.
7.2 Recovery Test
Annex 1 enthält keine expliziten Anforderungen für eine Nachweismethode der Clean Up Period. Deshalb ist es naheliegend, auf die ISO Standards zu verweisen, speziell auf den Abschnitt B.12, Recovery Test, in ISO 14644-3.
In der technischen Literatur wird der Begriff Recovery Time bzw. Erholzeit gleichbedeutend mit Clean Up Period aus dem regulatorischen Sprachgebrauch verwendet. Der Verdünnungseffekt der Lüftungsanlage für Reinräume mit turbulenter Mischströmung wird mit Hilfe der sog. Recoveryfunktion dargestellt:
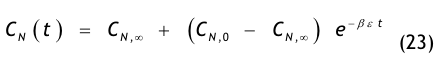
Eine kurzzeitige Kontaminierung des Reinraumes führt zu einer Erhöhung der Partikelanzahlkonzentration auf den Wert CN,0, der durch den Verdünnungseffekt nach einer gewissen Zeit wieder auf den Ruhewert CN,∞ abgebaut wird. Diese Dekontamination erfolgt exponentiell mit der Zeit t. Die Abbau geschwindigkeit bzw. Recovery Rate hängt von der Zerfallskonstante n = β ε ab, also dem Produkt aus Luftwechsel β und Lüftungseffektivität ε .
Damit die Reinheitsklasse at rest (CN,ar) in der geforderten Erholzeit auch sicher erreicht wird, muss der stationäre Endwert der Partikelanzahlkonzentration im Raum (CN,∞ ) deutlich unterhalb von CN,ar liegen. Dies wurde durch den sog. Antriebsfaktor δ = CN,ar / CN,∞ gekennzeichnet. Der Wert von CN,∞ wird bestimmt durch die inneren Partikelquellen (Personen, Equipment, Wirk- und Hilfsstoffe, Verfahrensabläufe), durch das gewählte Filterkonzept und durch die Anzahlkonzentration und Partikelgrößenverteilung des Außen luftaerosols. Die dazu erforderlichen Schätz methoden sind bekannt.
Die Auswertung der Erholzeitmessung sollte nach dem gezeigten Verfahren der vollständigen Regression (least squares approximation) erfolgen, insbesondere dann, wenn es nicht möglich ist, zwei Dekaden der Partikelanzahlkonzentration durchzumessen. Aus der vollständigen Regression erhalten wir die gesuchten Größen CN,∞ (und damit auch δ ) und n. Die Erholzeit wird dann rein rechnerisch ermittelt:
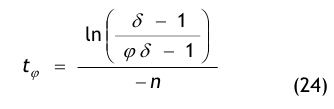
Eine Auswertung nach dem ISO-Verfahren, also unter Vernachlässigung von CN,∞ ≈ 0, führt auf eine Zerfallskonstante n*, die in der Regel kleiner ist als die aus der vollständigen Regression: n* < n. Dadurch wird auch die Erholzeit länger:
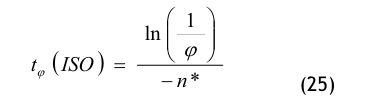
In vielen Fällen wird die Clean Up Period überschritten. Wenn dies im Rahmen der Qualifizierung einmal dokumentiert ist, müssen zwangsläufig erhebliche Korrekturmaßnahmen eingeleitet werden, die eigentlich überflüssig sind.
Thesen
Seit mehr als 15 Jahren verfolgen wir von Dohm Pharmaceutical Engineering die kontinuierliche Veränderung der cGMP-Anforderungen im wesentlichen der Regulierungsbehörden in Europa (EMEA) und den USA (FDA). Bei der Beratung unserer pharmazeutischen Kunden sehen wir täglich, welche schwierigen Randbedingungen bei der Umsetzung und Erfüllung dieser Anforderungen auftauchen. Als Dienstleister für die Pharmazeutische Industrie wollen wir nicht nur diese Randbedingungen aufzeigen, sondern gerade Lösungen hierzu anbieten. Deshalb beteiligen wir uns an der Diskussion zu den aktuellen proposals for amendment zum Annex 1 in Form von Einsprüchen bzw. durch diese vorliegende Ausarbeitung, deren Ergebnisse wir in den folgenden Thesen kurz zusammenfassen.
These 1:
In Reinräumen der Reinheitsklasse B gemäß Annex 1, in denen eine Freispülzeit tφ ≤ 20 min über zwei Dekaden ( φ = 100:1) nachgewiesen werden muss, liegen die erforderlichen Zuluft Raumluftwechsel ( βh) bei Werten 20/h, da man im allgemeinen von Spülgraden zwischen 0,75 und 0,9 ausgehen kann.
These 2:
In Reinräumen der Reinheitsklasse C kann der geforderte Zuluft-Raumluftwechsel (βh) bei Verwendung von HEPA-Filtern durchaus bei < 20/h liegen, da hier nur eine Freispülzeit tφ 20 min über eine Dekade (φ = 10:1) nachgewiesen werden muss, s. Abb. (7).
ACHTUNG: Für FDA-regulierte Produktionsbereiche (Supporting Clean Area Class 100.000) muss nach wie vor ein Zuluft-Raumluftwechsel von βh ≥ 20/h nachgewiesen werden.
These 3:
Der Richtwert für Erholzeiten von 15 bis 20 min, wie er noch immer durch den Annex 1 festgelegt ist, sollte auf „≤ 20 min“ geändert werden. Einen Richtwert als „von … bis“ zu umschreiben, führt zur Verwirrung. Er suggeriert, dass Erholzeiten von < 15 min nicht akzeptiert werden könnten.
Unser Textvorschlag für den neuen Annex 1 lautet daher:
» Die Grenzwerte für die Partikelanzahlkonzentration, die in der Tabelle für den Ruhezustand festgelegt sind, müssen nach einer kurzen Freispülphase von maximal 20 Minuten (Richtwert) wieder erreicht werden, und zwar ohne Anwesenheit von Personen und nach Abschluss aller Betriebstätigkeiten. «
These 4:
Bei Reinräumen der Reinheitsklasse D ist der Grenzwert für die in-operation-Konzentration im Annex 1 nicht festgelegt worden. Dadurch kann der Verdünnungsfaktor φ nicht bestimmt werden.
Wir empfehlen, den Verdünnungsfaktor auf φ = 2:1 festzulegen, sofern nicht andere pharmazeutisch, technologische Randbedingungen des Herstellprozesses bestehen. Daraus folgt, dass die Partikelanzahlkonzentrationen in operation die folgenden Werte annehmen:
- für Partikel ≥ 0,5 μm CN,io = 200.000 /ft³ bzw. 7.000.000 /m³,
- für Partikel ≥ 5,0 μm CN,io = 1.400 /ft³ bzw. 40.000 /m³.
These 5:
Wir empfehlen, dass auch in Reinräumen der Reinheitsklasse D HEPA-Filter verwendet werden.
Eine Untersuchung von ZIEMER und SCHENDERLEIN [17, 18] hat gezeigt, dass eine Außenluft Filtration nur mit Feinstaubfiltern der Güte F7 und F9 ohne Verwendung von endständigen HEPA-Filtern nicht ausreicht, um auf Dauer eine Reinheitsklasse von D at rest zu gewährleisten. Der stationäre Endwert der Raumkonzentration CN,∞ hängt wesentlich von der Planung ab: Filterkonzept, Konzentration und Partikelgrößenverteilung des Außenluftaerosols, Außenluftanteil, Partikelstrom und Partikelgrößenverteilung der inneren Quellen.
These 6:
Der Zuluft-Raumluftwechsel in Reinräumen der Reinheitsklasse D sollte mindestens 8/h bis 10/h unter Verwendung von endständigen HEPA Filtern betragen, s. Abb. (8).
These 7:
Mit Hilfe der o. g. Auslegungskriterien für den Luftwechsel und die damit verbundene Erholzeit kann man auch Schleusensysteme statt als Mehrkammer- auch als Einkammer-Schleusen ausbilden. Dabei müssen der Luftwechsel, die Lüftungseffektivität und die Erholzeit nach einer definierten Belastung (Umkleideprozedur etc.) so validiert werden, dass der Nachweis für die gleiche Reinheitsklasse der Schleuse gegenüber dem Reinraum erbracht wird.
Autoren

Dipl.-Ing. (TU) Thomas RAATZ hat Verfahrenstechnik an der TU Dresden studiert und ist seit mehr als 12 Jahren für die Pharmazeutische Industrie im Bereich der Qualifizierung von reinraumtechnischen Anlagen tätig, davon die letzten 6 Jahre als Projektleiter bei Dohm Pharmaceutical Engi neering.

Dr.-Ing. Wolf ZIEMER hat Energie- und
Verfahrenstechnik an der TU Berlin studiert und an
der TU Budapest über das Thema Aerosolfiltration
promoviert. Er ist seit 25 Jahren im Bereich
Reinraumtechnik und Pharmazeutische Verfah
renstechnik tätig, davon die letzten 6 Jahre bei
Dohm Pharmaceutical Engineering.
Tabellen
Tabelle 1
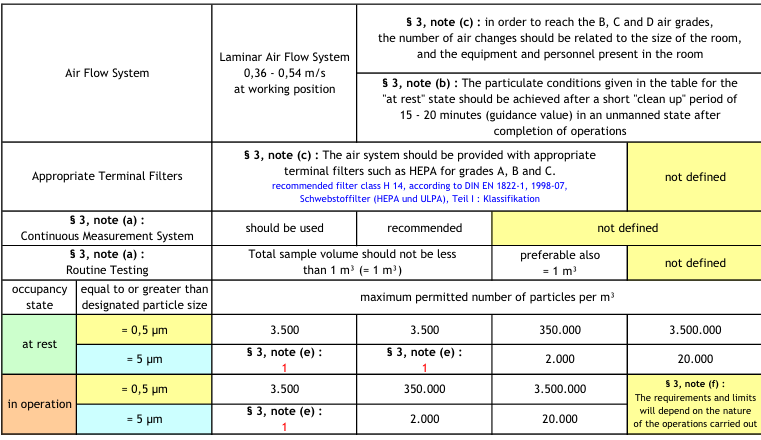
Tabelle 2
Übersicht über die vollständige Regression
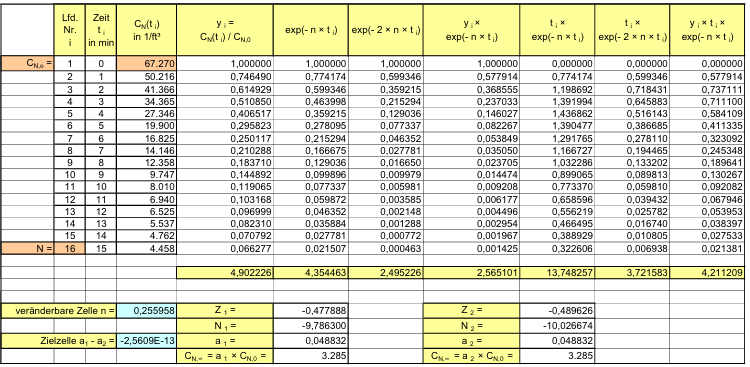
Abbildungen
Abbildung 2 Clean Up Period für einen B-Raum
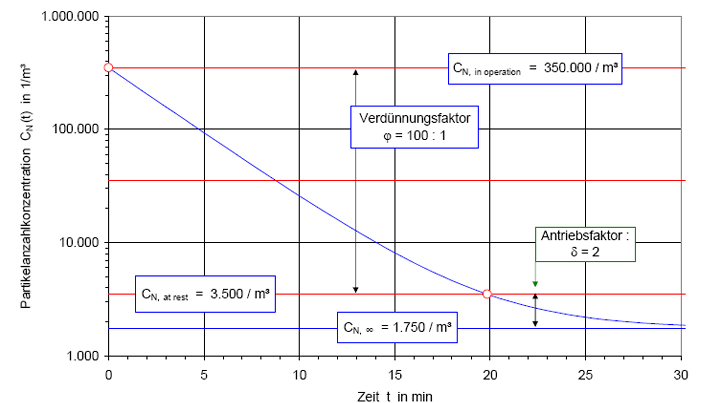
Abbildung 3 Beispiel Versuchswerte & Regressionskurve
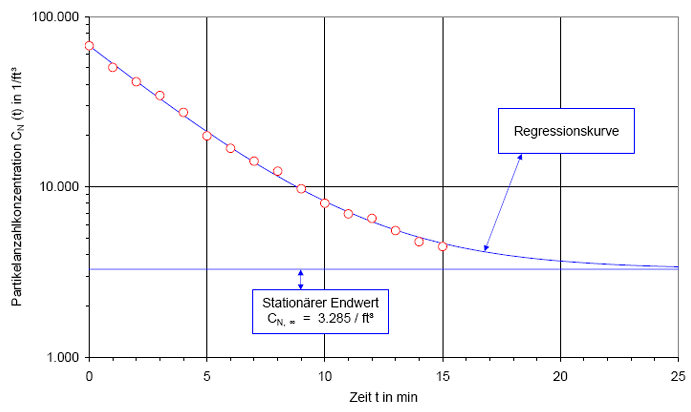
Abbildung 4 Beispiel Vergleich EXCEL-Regression und wahre Exponentialregression
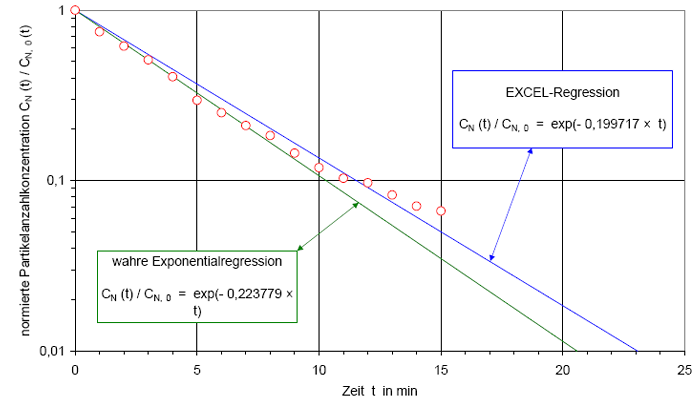
Abbildung 5 Beispiel vollständige Regression
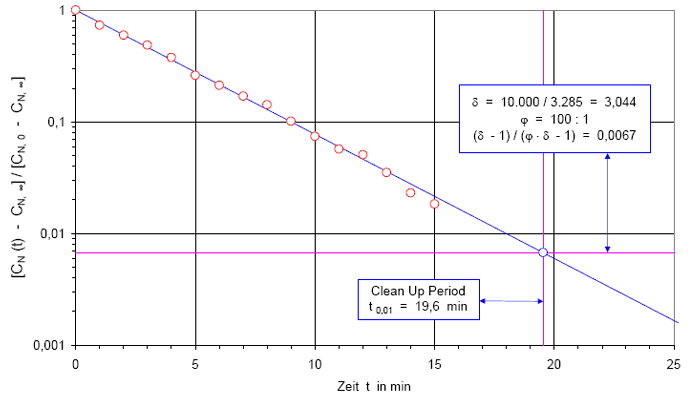
Abbildung 6 Luftwechsel für B-Räume mit HEPA-Filtern
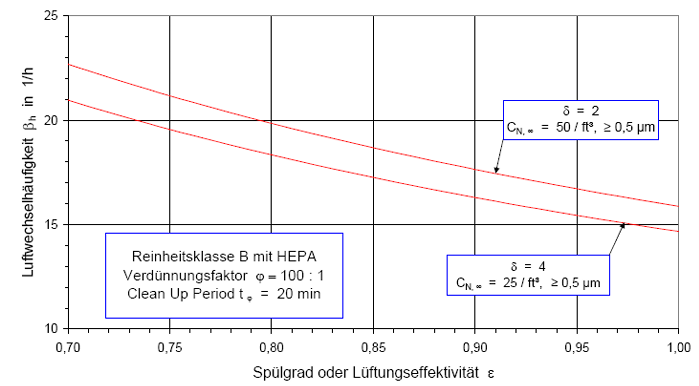
Abbildung 7 Luftwechsel für C-Räume mit HEPA-Filtern
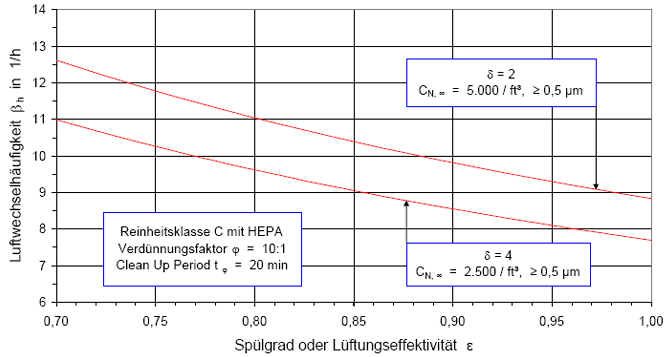
Abbildung 8 Luftwechsel für D-Räume mit HEPA-Filtern
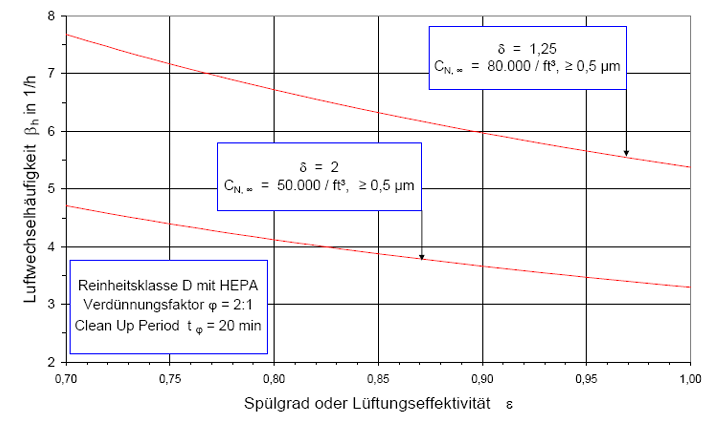
Anhänge



Anhang 2: Exponentialregression


Literatur
1
EG-Leitfaden einer Guten Herstellungspraxis für Arzneimittel, in Kraft getreten am 1. Januar 1992, zusammengestellt von Gert AUTERHOFF, ECV Editio Cantor Verlag, Aulendorf, 1990, als Sonderdruck aus: Die Pharmazeutische Industrie, Jhg. 52, Nr. 7, pp.: 853-883, 1990
2
EG-Leitfaden einer Guten Herstellungspraxis für Arzneimittel, zsgest. und hrsg. von Gert AUTERHOFF, 2. überarb. und erw. Aufl., ECV Editio Cantor Verlag, Aulendorf, 1993
3
EG-Leitfaden einer Guten Herstellungspraxis für Arzneimittel, zsgest. und hrsg. von Gert AUTERHOFF, 3. überarb. und erw. Aufl., ECV Editio Cantor Verlag, Aulendorf, 1994
4
EG-Leitfaden einer Guten Herstellungspraxis für Arzneimittel, zsgest. und hrsg. von Gert AUTERHOFF, 4. überarb. und erw. Aufl., ECV Editio Cantor Verlag, Aulendorf, 1995
5
EG-Leitfaden einer Guten Herstellungspraxis für Arzneimittel, zsgest. und hrsg. von Gert AUTERHOFF, 5. überarb. und erw. Aufl., ECV Editio Cantor Verlag, Aulendorf, 1998
6
EG-Leitfaden einer Guten Herstellungspraxis für Arzneimittel, zsgest. und hrsg. von Gert AUTERHOFF, 6. überarb. und erw. Aufl., ECV Editio Cantor Verlag, Aulendorf, 2000
7
EC Guide to Good Manufacturing Practice, (http://pharmacos.eudra.org/F2/eudralex/vol 4/home.htm),Revision to Annex 1: Manufacture of Sterile Medicinal Products, adopted by Pharmaceutical Committee, May 2003, in operation September 2003
8
http://www.emea.eu.int/Inspections/ WhatsNew.html: What is NEW in Inspections Sector, Mitteilung vom 23.11.2005: Good Manufacturing Practice Annex 1: Proposals for Amendment to the Environmental Classification Table for Particles and Associated Text, London, 21. September 2005
9
http://www.fda.gov/cder/guidance/index.html: Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice, September 2004
10
DIN EN ISO 14644-3, Entwurf, Dezember 2002: Reinräume und zugehörige Reinraumbereiche, Teil 3: Messtechnik und Prüfverfahren, 70 Seiten
11
ISO / FDIS 14644-3:2004(E = English): Cleanrooms and associated controlled environments, Part 3: Test methods, prepared as Document N 161by ISO TC 209/SC/WG 3, 2004-11-5
12
ISO 14644-3:2005(E): Cleanrooms and associated controlled environments, Part 3: Test methods, first edition, December 15, 2005
13
VDI 2083-2:1996: Reinraumtechnik, Bau, Betrieb und Instandhaltung, 27 Seiten
14
RYDBERG, J. und KULMAR, E.: Lüftungseffekt bei verschiedenen Plazierungen der Lufteinströmungs- und Luftausströmungsöffnungen, Installation, Vol. 19, pp.: 165-170, 1947.
15
Max von PETTENKOFER: Ueber Ventilation od. Luftwechsel in Wohngebäuden, Münchener Kalender für das Jahr 1886
16
LINGNAU, J. et al.: EEC-Guide to Good Manufacturing Practice for Medicinal Products/Supplementary Guidelines/Manufacture of Sterile Medicinal Products: Air Classification System, Diskussion der Reinheitsklassen-Anforderungen, Pharmazeutische Industrie, Bd. 51, Heft 12, pp.: 1380-1384, 1989
17
ZIEMER, W.; SCHENDERLEIN, S.: Particle Size Distribution in Pharmaceutical Classrooms, Environmental Engineering, Vol. 7, No. 3, pp.: 22-29, 1994
18
ZIEMER, W.; SCHENDERLEIN, S.: Particle Size Distribution in Pharmaceutical Classrooms, Proceedings of the 12th ISCC (International Symposium on Contamination Control), pp.: 79 86, 10.-14. October 1994, Pacific Convention Plaza, Yokohoma, Japan
1 Einleitung
1.1 Aufgabenstellung
Im Rahmen der Qualifizierung (OQ) sollte die Integrität der HEPA Filter der Filterklasse H 13 und die Reinheitsklasse in einem Heißluft Sterilisationstunnel im kalten Zustand (23C) gemessen werden.
Es handelt sich dabei um einen Heißluft Sterilisation Tunnel für die Sterilisierung und Depyrogenisierung von Vials.
In diesem Report soll nun die Frage diskutiert wer den, ob es erforderlich und sinnvoll ist, die Reinheitsklasse auch in operation, also im heißen Zu stand (240C) nachzuweisen. Die Reinheit im Tunnel hängt im wesentlichen von dem Rückhaltevermögen der Filter und der Güte der Probenahme im heißen Zustand ab:
- 1. Wie verhält sich ein HEPA Filter bei höheren Temperaturen?
- 2. Welchen Einfluss übt die höhere Temperatur auf die Probenahme aus?
1.2 Begriffe
1.2.1 Definition: at rest (kalt)
Die Reinheitsklassen werden im Annex 1, EG Leitfaden für zwei Betriebszustände definiert: at rest und in operation.
Die Reinheitsklassen werden im Annex 1, EG Leitfaden für zwei Betriebszustände definiert: at rest und in operation.
Für den Nachweis der Reinheitsklasse in einem Heißluft-Sterilisationstunnel bedeutet der Betrieb zustand at rest aus unserem Verständnis eine Prüfung im kalten Zustand, d.h. es befinden sich keine Vials auf dem laufenden Transportband, die Ventilatoren für die Luftversorgung des Tunnels laufen, aber die Heizung für den Heizteil ist abgeschaltet.
1.2.2 Definition: in operation (heiß)
Für den Nachweis der Reinheitsklasse in einem Heißluft-Sterilisationstunnel bedeutet der Betrieb zustand in operation aus unserem Verständnis eine Prüfung im heißen Zustand, d.h. es befinden sich keine Vials auf dem laufenden Transportband, die Ventilatoren für die Luftversorgung des Tunnels laufen und die Heizung ist für den Heizteileinge schaltet.
1.3 Ziel
Das Ziel dieses Berichtes besteht darin, den Nachweis darüber zu führen, dass die Reinheitsklasse im Heißluft-Sterilisationstunnel im Betriebszustand at rest (kalt) gemessen werden sollte. Dafür sprechen zwei Gründe:
- 1.Der Abscheidegrad der HEPA Filter steigt bei höheren Temperaturen und verbessert da durch die Reinheitsklasse.
- 2. Bei höheren Temperaturen nimmt auch die Abscheidung der Aerosolteilchen im Ansaug system des Partikelzählers aufgrund der verstärkten Thermophorese zu. Dadurch ist der Nachweis der Reinheitsklasse im Betriebszustand in operation (heiß) mit größeren Messunsicherheiten behaftet als im kalten Zustand.
2 Filtercharakteristik
2.1 Penetrationskurve und MPPS
Unter der Filtercharakteristik versteht man den Funktionsverlauf der Penetration in Abhängigkeit von der Partikelgröße. Kleine Aerosolteilchen werden aufgrund der Brownschen Bewegung (Einsteinsche Diffusion) abgeschieden, große Teilchen werden aufgrund des sog. Sperreffektes und ihrer Trägheit abgeschieden. Aerosolteilchen, die bereits zu groß sind, um noch wirksam durch Diffusion abgeschieden zu werden, aber nicht groß genug sind, um wirksam durch Trägheitseffekte abgeschieden zu werden, weisen den größten Durchgang auf, MPPS genannt: Most Penetrating Particle Size oder Partikelgröße maximaler Penetration.
2.2 MPPS-Prüfung
Die Penetrationskurve sieht aus wie eine umgekehrt liegende Parabel und besitzt ein ausgeprägtes Maximum im MPPS. Genau an dieser Stelle wird nach DIN EN 1822 das Filter geprüft und klassifiziert. Die Lage des MPPS hängt von weiteren Parametergrößen des Filtermediums ab, z.B.:
- mittlere Faservolumendichte oder Packungs dichte
- Blattdicke des Filtermediums (caliper)
- Faserdurchmesser bzw. Fasergrößenverteilung
- Anströmgeschwindigkeit zum planen Filter medium
- Temperatur der Luft (Dispersionsmedium)
- Umgebungsdruck
2.3 Berechnung der Penetration
Seit mehr als 70 Jahren wird die Filtertheorie kontinuierlich ausgebaut. Unter Filtertheorie wird die mathematisch-physikalische Theorie der Filtration von Aerosolen in Faserfiltermedien verstanden. Mit Hilfe geeigneter Struktur- und Strömungsmodellen kann der Abscheidegrad bzw. die Penetration der Aerosolteilchen in Faserfiltermedien berechnet und der Einfluß der o.g. Parameter untersucht werden. Das von uns benutzte Filtermodell ist in den 1970 er Jahren von Fuchs, Stechkina und Kirsch [1] entwickelt worden und wird nachfolgend FSK Modell genannt.
2.4 HEPA-Filter H 13
2.4.1 Technische Daten
Im Heizteil eines Heißluft-Sterilisationstunnels wurde hier beispielweise ein H 13 Filter der Fa. Camfil angenommen, Modell bzw. Filterbezeichnung: 1FRKV-725-1W. Solche temperaturbeständigen HEPA-Filter sind meist nur in der Qualität H 13 erhältlich. Das Filtermedium besteht meist aus Quarzglasfasern ohne Bindemittel, das bei den hohen Temperaturen ohnehin verdampfen würde. Zur Versteifung der Konstruktion und Separation der Plisseefalten werden gewellte Metallseparatoren verwendet, die beständig gegen die hohen Temperaturen sind.
Die folgenden technischen Daten wurden aus dem Camfil-Prospekt (Absolute 1FRK) bzw. dem Filtertestbericht entnommen:
- Abmessungen B H T:610 457 292 jeweils in mm und als Außenmaße des Filterrahmens
- Filterfläche AF : 16,4 m2
- Volumenstrom, nominal Vnom: 1500 bzw. 1420 m3/h
- Druckdifferenz: 250 Pa
- Geschwindigkeit zum planen Filtermedium:
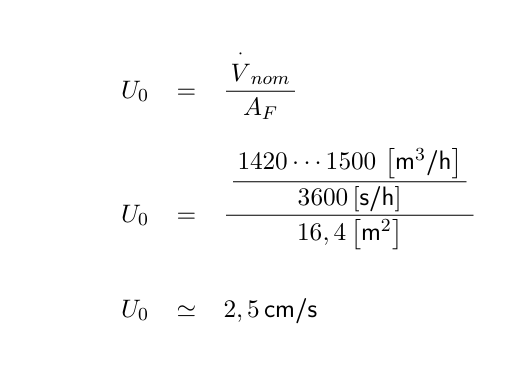
2.4.2 Ergebnis
Bei normaler Umgebungstemperatur von 23C wird die Filtercharakteristik für das H 13 Filter ausgedrückt durch einen Abscheidegrad von 99,95 % im MPPS bzw. eine maximale Penetration (Pmax) von 0,05 % bzw. absolut 5×10-4 im MPPS:
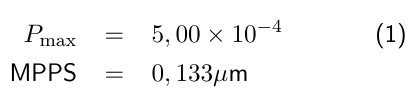
Bei einer Temperatur von 240C steigt der Abscheidegrad bzw. sinkt die Penetration aufgrund der verbesserten Diffusionsabscheidung und daraus erhält man nach dem FSK-Modell die folgende Filtercharakteristik:

Wie man sieht, verringert sich die Penetration bei 240°C gegenüber der Penetration bei 23°C um den Faktor 6. Die Angaben über den MPPS bei 0,3 µm in den Filtertestberichten der Camfil können nicht korrekt sein. Der berechnete MPPS liegt bei 0,133 bis 0,158 µm.
3 Nachweis der RH Klasse
3.1 Messung im Raum
Der Nachweis der Reinheitsklasse erfolgt nach der Methode, die in der ISO 14644-1 beschrieben ist. Man benutzt einen Optischen Partikelzähler (kurz OPZ), mit dem die Partikelanzahlkonzentration in den Reinräumen gemessen wird. Diese Messung erfolgt bei den üblichen Umgebungstemperaturen. Auch sind alle Orte, an denen man die Reinheitsklassen nachzuweisen hat, einfach zugänglich, sodaß keine Ansaugsysteme in Form von Ansaugschläuchen erforderlich sind.
3.2 Messung im kalten Tunnel
Die Besonderheiten, die beim Nachweis der Reinheitsklasse in einem Heißluft-Sterilisationstunnel zu beachten sind, bestehen darin, dass nunmehr ein Ansaugsystem erforderlich wird. Um die entfernteren Orte im Tunnel zu erreichen, braucht man einen Ansaugschlauch mit einer Länge von mehr als 3 m. Üblicherweise benutzt man zur Verbindung der isokinetischen Ansaugsonde mit dem Partikelzähler einen sog. Getränkeschlauch (Bev-A Line), der im Innern sehr glatt und elektro-statisch ableitfähig ist, um eine Abscheidung der Aerosolteilchen aufgrund elektrischer Ladung zu vermeiden, oder wenigstens zu minimieren. Dennoch kann man eine Abscheidung der Teilchen nicht vermeiden. Im Schlauch herrscht eine turbulente Rohrströmung und die Turbulenz verursacht eine turbulente Querbewegung und eine Trägheitsabscheidung der Teilchen an die Rohrinnenoberfläche.
Für Teilchen der Größe 0,5 µm ist diese Abscheidung vernachlässigbar, für die der Größe 5 µm allerdings erheblich je länger der Schlauch ist. Da durch entsteht eine Messunsicherheit, denn es können ja nur die Teilchen gezählt werden, die das Sichtvolumen (view volume) des Partikelzählers auch erreichen.
3.3 Messung im heißen Tunnel
Wenn die Anforderung gestellt wird, die Reinheitsklasse im Heizbetrieb zu messen, also in operation, muss das Ansaugsystem verändert werden. Wegen der hohen Temperaturen muss ein Ansaugrohr aus Edelstahl genommen werden, das mit einer Mantelkühlung versehen ist. Die Kühlung der Probenahmeluft dient zum Schutz des Partikelzählers. Bei einer Temperatur von 240C würden alle optischen und elektronischen Bauteile im OPZ zerstört.
Durch die Kühlung entsteht ein Temperaturgradient, d.h. die Temperatur der Probenahmeluft in unmittelbarer Nähe zur inneren Wandoberfläche des Probenahmerohres ist deutlich höher als die der Wandoberfläche selbst. Dieser Temperaturgradient verursacht eine Thermodiffusion, auch Thermophorese genannt, und treibt die Teilchen an die kalte Rohroberfläche. Die Abscheidung der Teilchen wird verstärkt.
4 Zusammenfassung
- 1. Es gibt keine dezidierte regulatorische Anforderung, die einen Nachweis der Reinheitsklasse im heißen Zustand für Heißluftsterilisatoren (o.ä.) empfiehlt oder vorschreibt.
- 2. Ein Nachweis der Reinheitsklasse im Heißlufttunnel in operation bringt keinen Vorteil, weil der Abscheidegrad der HEPA-Filter bei höheren Temperaturen ansteigt und die Reinheitsklasse besser wird bzw. dadurch die Partikelanzahlkonzentration im Tunnel sinkt.
- Durch die Kühlung des Probenahmesystems, die bei einem Nachweis in operation (heiß) zum Schutz des Partikelzählers erforderlich wird, ist mit einem erheblichen Partikelverlust aufgrund der Thermodiffusion zu rechnen. Teilchen, die den OPZ nicht erreichen, können nicht gezählt werden und stellen eine große Messunsicherheit dar bzw. stellen den Nachweis der Reinheitsklasse überhaupt in Frage.
- Der Nachweis der Reinheitsklasse at rest (kalt) stellt die strengere Prüfung dar und ist deshalb zu empfehlen.
Anhang
Filtercharakteristik am Beispiel eines Heißluft-Sterilisationstunnels
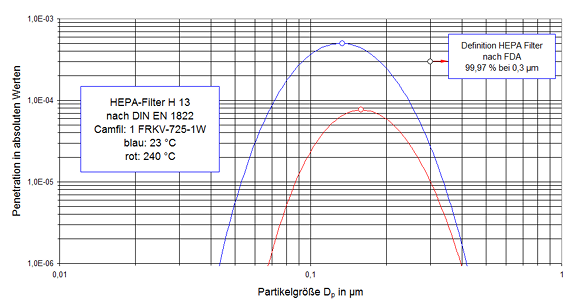
Literatur
1
[1] Kirsch, A. A. and Stechkina, I. B.: The Theory of Aerosol Filtration with Fibrous Fil ters, Chapter 4, pp.:165-256, in Fundamantals of Aerosol Science, ed. D. T. Shaw, John Wi ley & Sons, New York, 1978
1 Einleitung
Die Wirkung des Windes auf Bauwerke ist in den letzten Jahrzehnten eingehend untersucht worden, siehe [1]. Bei einem schweren Sturm mit Windgeschwindigkeiten von 100- 150 km/h entsteht auf der Luvseite eines Gebäudes ein Staudruck von 500- 1 000 Pa. Diese Kräfte müssen natürlich in der Statik des Gebäudes und bei der Gestaltung der Bauwerksteile (z. B. Fassadenelemente) berücksichtigt werden.
Die Windauflastung führt aber auch zur Infiltration, d.h. zum Eindringen der Außenluft durch die Bau werksfugen und damit zur natürlichen Durchlüftung des Bauwerks. Bei der Auslegung von Heizungs und Klimaanlagen wird die Infiltration durch die Berechnung des Lüftungswärmebedarfs berücksichtigt. Bei Gebäuden, in denen pharmazeutische Produkte hergestellt werden, verursacht die Infiltration zusätzlich eine Kontamination, denn mit der eindringenden Außenluft werden die darin dispergierten Aerosolteilchen in das Gebäude eingeschleppt. Um die Reinräume gegen diese Kontamination durch Infiltration zu schüt zen, wird häufig eine Raum-in-Raum-Bauweise gewählt, d.h. den Reinräumen werden z.B. umlaufende „schwarze“ Gänge vorgelagert, die eine Schutzbarriere gegen den direkten Partikeleinbruch bilden. Wegen der Knappheit der Baufläche werden in zunehmendem Maße die Reinraumflächen bis an die Außenwand ausgedehnt und genutzt. Dadurch wird die Außenwand zur Reinraumwand mit besonders hohen bauphysikalischen Anforderungen: Ausdehnungsfreiheit wegen der thermischen Lastwechsel ( ΔT = 60-80 K), hohe Fugendichtigkeit auch bei extremer Windauflastung, hohe Diffusionsdichtheit und gute Wärmedämmung, damit keine Kondensation und Schimmelbildung auf der Wandinnenseite stattfindet.
In diesem Aufsatz soll speziell der Partikeltransport durch Fassadenfugen und sein Einfluss auf die Rein heit betrachtet werden. Die Frage lautet: wie dicht muss eine Fassade sein, damit im Reinraum eine Reinheitsklasse C (in operation) nach EG-Leitfaden, Annex 1 [15] auf Dauer sicher eingehalten wird?
Wir haben einen Raum von LR BR = 10 m 10 m= 100 m2 Fläche mit einer Höhe von HR = 3 m als Modell angenommen. Das Modellraumvolumen beträgt somit 300 m3. Bei einem Zuluft-Raumluftwechsel von β=20 1/h beträgt der Zuluft-Volumenstrom ˙ Vzu = 6 000 m3/h. Eine Wandfläche von 10 m X 3 m = 30 m2 soll eine Außenwand sein, durch die bei einer Windauflastung von 500 Pa der Infiltrationsstrom ˙ VINF = μVZU durchfließen kann, wobei = Migrationskoeffizient.
Die der Modellgleichung zugrunde gelegte RLT-Anlage ist in Abb. (1) dargestellt. Sie besteht aus einem Außenluft-Vorbehandlungsgerät und einem Umluftgerät. Da wir ausschließlich den Partikeltransport betrachten, sind im Anlagenschema lediglich die Filterstufen eingezeichnet. Es wird angenommen, dass die in den Geräten vorhandenen Bauelemente (speziell die Ventilatoren und Luftbefeuchter) selbst keine Partikelproduzenten sind. Die im Schema eingezeichneten 8 Filterstufen (3 im Außenluftgerät, 3 im Umluftgerät und je 1 endständiges Filter in der Zuluft bzw. Abluft) sollen lediglich Positions- und Bestückungsvarianten darstellen. Für die Entwicklung der Berechnungsgleichung (22) war es erforderlich, alle Varianten (Platzhalter oder Dummy-Positionen) aufzunehmen. Für die rechnerische Simulation einer
2. Aerosolphysikalische Grundlagen
realen Anlage werden alle nicht benötigten Filtervarianten dadurch eliminiert, dass die entsprechende Penetration Pj auf Eins gesetzt wird. Ein Filter mit der Penetration P = 1 lässt alle Teilchen hindurch, d.h. es ist rechnerisch nicht vorhanden. In den anschließenden Abschnitten (2 bis 6) werden die Grundlagen erläutert, die für die Aufstellung der Berechnungsgleichung erforderlich sind.
2.1 Anzahlkonzentration und Partikelgrößenverteilung
Anzahlkonzentration bzw. Anzahldichte und Partikelgrößenverteilung (PGVT) sind fundamentale Größen der Aerosolphysik zur Beschreibung der Eigenschaften von Partikelkollektiven. Es ist deshalb sehr erstaun lich, dass diese Grundbegriffe in der Literatur nicht adäquat definiert werden, siehe z.B. [2, 3, 4]. Nach Ziemer [5] muss man folgende Begriffe unterscheiden:
1.lokale, kumulative Anzahlkonzentration

2. lokale, fraktionelle Anzahlkonzentration

3. Partikelgrößenverteilung (PGVT)
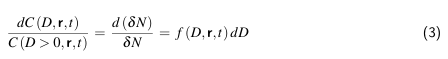
Die Größen bedeuten: δN = Anzahl aller Teilchen der Größe D > 0, die zur Zeit t am Ort r in δV dispergiert sind. Das Dispersionsmittel wird als Kontinuum betrachtet, das als solches keine diskrete Struktur besitzt. Dann sind alle Teilchen mit der Eigenschaft D > 0 prinzipiell abzählbar. δV = elementares Volumenelement um den Ort P(r) aus dem Kontrollvolumen (KV) V, klein genug um die Anwendung der Infinitesimalrechnung zu rechtfertigen, aber doch so groß, dass es eine genügende Partikelzahl δN enthält, um eine Partikelgrößenstatistik gemäß Gl. (3) betreiben zu können. d( δN) = monodisperse Fraktion von δN, d.h. Anzahl der Teilchen des Größenintervalls [D; D + dD], die zur Zeit t am Ort r in δV dispergiert sind. f(Dr t) = Häufigkeitsdichte der PGVT nach der Mengenart „Anzahl“.
2.2 2.2 Erhaltungssatz und Partikelstrom
Gl. (2) wird nach d(δN) umgestellt und anschließend wird über das KV integriert:
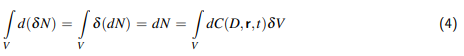
Bei der Integration der linken Seite von Gl. (4) haben wir die kommutative Eigenschaft der Operatoren „d“ und „δ“ benutzt. Von Gl. (4) wird nun die substantielle Ableitung gebildet:
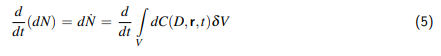
wobei dN/dt = N˙ = kumulativer Partikelstrom für Teilchen mit D > 0 und d(dN/dt) = dN˙ = Fraktionsteilchenstrom oder fraktioneller Partikelstrom für Teilchen aus dem monodispersen Partikelgrößenbereich [D; D + dD]. Der fraktionelle Partikelstrom in Gl. (5) wird noch nach Quellen- und Senkenstrom aufgespalten: dN˙ = dN˙q − dN˙s . Damit lautet der Erhaltungssatz der Partikelanzahl:

Die linke Seite von Gl. (6) beschreibt den rein konvektiven Transport von Teilchen zu/aus dem KV. Die rechte Seite von Gl. (6) hat die Bedeutung verallgemeinerter Quellen- und Senkenströme, d.h.:
- a) Zu- / Abfluss von Teilchen der monodispersen Fraktion [D; D + dD] über die Grenzen des KV’s aufgrund nicht konvektiver Erscheinungen (Trägheit, Sedimentation, Diffusion aufgrund der Brownschen Bewegung, etc.)
- b) Zu- / Abnahme der Teilchenzahl einer betrachteten Fraktion [D; D + dD] im KV durch Koagulation, Kondensation oder Verdampfung
- c) Partikelquellen und -senken im KV, die als diskrete Generatoren bzw. Adsorber vorliegen können oder auch quasi-kontinuierlich im KV verteilt sein können.
Die substantielle Ableitung nach Euler lautet:
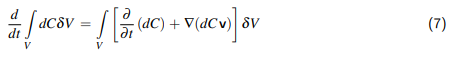
wobei v = Vektor der Geschwindigkeit des Dispersionsmittels
2.3 Partikelstrombilanz
Wir betrachten den Spezialfall einer stationär durchströmten Stromröhre. Wegen der Stationärität ist ∂(dC)∂ (t) = 0. Der übrigbleibende Teil des Volumenintegrals wird mit Hilfe des Integralsatzes von Gauss in ein Flächenintegral umgewandelt:
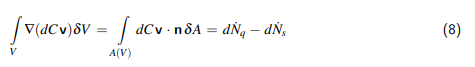
wobei n = der nach außenweisende Einheitsvektor der Flächennormalen ist. Für die einfache Stromröhre kann das Flächenintegral mit den folgenden Annahmen berechnet werden:
- a) die Querschnitte A1 und A2 stehen senkrecht zur Achse der Stromröhre, d.h. die Vektoren v1 und n1 bzw. v2 und n2 verlaufen parallel zueinander, so dass v1 · n1 = −v1 und v2 · n2 = +v2. Auf dem Mantel der Stromröhre gilt vO · nO = 0
- b) Die fraktionelle Anzahlkonzentration, PGVT und Geschwindigkeit sind konstant auf den Querschnitten A1 und A2.
Damit erhalten wir
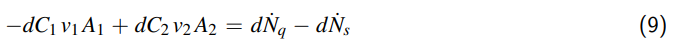
Die Größen vj Aj = V˙ j stellen bekanntlich Volumenströme dar. Für die stationäre Strömung in einer allgemeinen Stromröhre mit vielen Ein- und Austrittsflächen gilt dann:
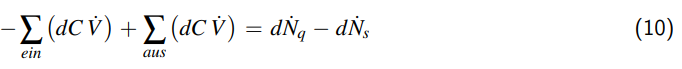
Die spätere Berechnungsgleichung basiert auf der Bilanz von Fraktionsteilchenströmen, die mit Hilfe von Gl. (3) in folgender Form geschrieben werde:
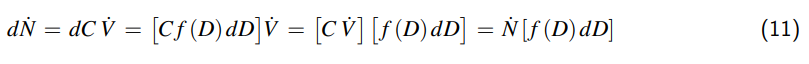
Partikelströme durch Kanäle und Räume können mit der o. a. Stromfadentheorie näherungsweise eindimensional behandelt werden, wenn man für die Geschwindigkeit, Anzahlkonzentration und PGVT über den Querschnitt gemittelte Werte benutz
3 Das atmosphärische Aerosol
Das atmosphärische Aerosol beeinflusst die Reinheit des Reinraumes auf zweierlei Weise:
- a) Außenluft wird von der RLT-Anlage angesaugt und das darin enthaltene Aerosol gelangt in den Reinraum, wenn auch in Anzahlkonzentration und PGVT durch die verschiedenen Filterstufen abgeschwächt,
- b) durch kleine Fugen in der Außenfassade dringt bei entsprechender Windauflastung die Außenluft ungefiltert in den Reinraum.
Um diese Einflüsse rechnerisch abschätzen zu können, ist es erforderlich, die Anzahlkonzentration und PGVT des atmosphärischen Aerosols zu kennen (möglichst auch den Gehalt an Mikroorganismen). Die ersten systematischen Messungen hat C. Junge im Frühjahr 1951 am meteorologischen Institut in Frankfurt am Main durchgeführt. Junge [6] hat von 25 Kontinentalaerosolen die Anzahlkonzentration und PGVT gemessen und eine bemerkenswerte Konstanz der Verteilung im Partikelgrößenbereich D = 0,2 … 10 µm festgestellt:
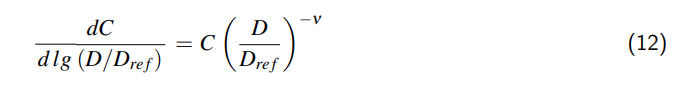
Die logarithmische Fraktionsdichte konnte durch eine Potenzverteilung dargestellt werden, wobei der Exponent ν ≅ 3 betrug. Diese Verteilung ist als Junge-Verteilung bekannt geworden. Heute wird das atmosphärische Aerosol meist durch eine tri-modale LNVT beschrieben entsprechend den 3 Komponenten (Modi), die man identifiziert hat:
- 1. NM = Nucleation Mode (Kernbildungsanteil), der aus ultrafeinen Teilchen der Größe D < 0,1 µm besteht mit hoher Anzahldichte
- 2. AM = Accumulation Mode (Koagulationsanteil), der aus submikroskopischen Teilchen der Größe D = 0,1 … 1 µm besteht mit mittlerer Anzahldichte
- 3. CM = Coarse Mode(Grobanteil), der aus Fein-und Grobstaubteilchen der Größe D > 1 µm besteht mit geringer Anzahldichte.
Die fraktionelle Anzahlkonzentration des atmosphärischen Aerosols wird somit wie folgt beschrieben:
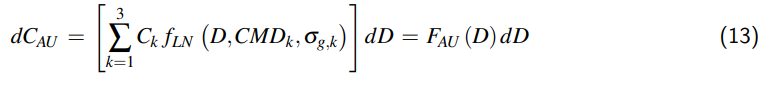
mit FAU (D) = Fraktionsdichte in 1/m4 oder 1/(cm3 µm) als Kurzschreibweise der in eckigen Klammern stehenden Formel.
Die LNVT Komponenten lauten wie folgt
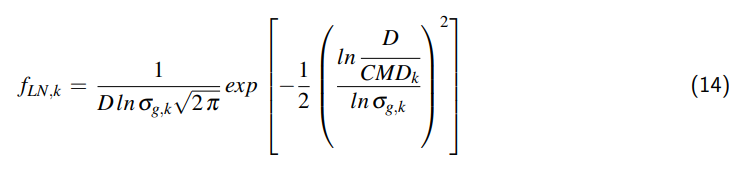
Jede Komponente k wird durch 3 Parameter beschrieben: Ck = kumulative Anzahlkonzentration, CMDk = Mediandurchmesser und σg,k = geometrische Standardabweichung.
Wir haben das von Junge gemessene Stadtaerosol von Frankfurt a. M. in eine tri-modale LNVT zerlegt und die 9 Parameter in Tab. (1) zusammengefasst.
4 Partikelquellen im Raum
Kontamination wird überwiegend durch den Menschen und seinen Aktivitäten verursacht. Wir haben uns deshalb auf die Partikelabgabe durch die im Reinraum tätigen Menschen konzentriert und die Partikelerzeugung der Produktionsmaschinen bzw. die der Prozesse vernachlässigt. Dies natürlich auch deshalb, weil keine zuverlässigen Angaben darüber vorliegen.
Über die Partikelabgabe des Menschen in Abhängigkeit des Aktivitätsgrades und des Typus der Reinraumbekleidung hat Minamino [7] eine Reihe vorzüglicher Messungen durchgeführt. Wir haben seine Werte nochmals überarbeitet und festgestellt, dass sie sich im Größenbereich D = 0,3 … 15 µm durch eine bimodale LNVT darstellen lassen, jeweils für geringe und hohe Aktivität.
Die geringe Aktivität wird definiert durch leichte Armbewegungen im Sitzen, die hohe Aktivität durch
Laufen, Aufstehen/Hinsetzen und Gymnastik. Die Reinraumbekleidung bestand aus einem Overall, den
Armschützern, den Überschuhen und einer Kopfhaube. Der Overall bestand aus fusselfreiem 100% Polyester und war elektrostatisch ableitfähig durch eingewebte Kohlefasen jeweils in 5 mm Abstand zueinander.
In Abb. (5) ist der kumulative Partikelstrom pro Person als Funktion der Partikelgröße dargestellt:
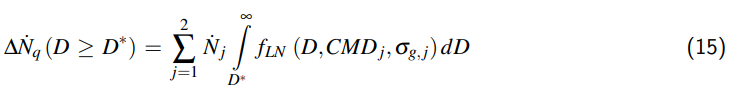
mit den beiden Aktivitätsgraden als Parameter. Die 2 x 6 Parameter (N˙j ,CMDj ,σg j) sind in Tab. (3) zusammengestellt. Aus der raumseitigen Partikelstrombilanz wird der vom Menschen produzierte Fraktionsteilchenstrom dN˙q in eine fraktionelle Anzahlkonzentration dCq umgewandelt:
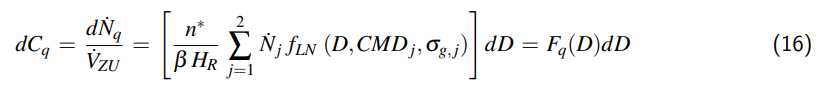
mit n* = Besetzungsdichte (Anzahl Personen pro Fläche), β = Zuluft-Raumluftwechsel, HR = Raumhöhe und Fq(D) = Fraktionsdichte der Partikelquellen im Raum.
5 Luftfilter
In der Reinraumtechnik werden zwei Arten von Luftfiltern verwendet: Standard-Luftfilter zur Vorfiltration, deren Filtrationsleistung nach DIN EN 779 [13], klassifiziert werden, und Schwebstoff-Filter nach DIN EN 1822-1 [14]. Beide Normen setzen unausgesprochen voraus, dass die Filtermedien aus Fasern bestehen.
Gemessen an dem heutigen Stand unserer Kenntnisse der Aerosolphysik, speziell der Filtertheorie, und der verfügbaren Aerosolmesstechnik weisen die beiden Filternormen große Unterschiede in der Prüfmethodik auf. Die Klassifizierung der Schwebstoff-Filter nach DIN EN 1822-1 basiert auf der fraktionellen Penetration nach der Mengenart „Anzahl“ im MPPS = Most Penetrating Particle Size, zu deutsch PGMP = Partikelgröße maximaler Penetration. Bei der Klassifizierung der Vorfilter nach DIN EN 779 muss man zunächst zwei Filterklassen unterscheiden: Grobstaubfilter (G1 – G4) und Feinstaubfilter (F5 – F9). Für die Reinraumtechnik kommen nur die Feinstaubfilter in Betracht. Diese werden nach DIN EN 779 mit DEHS beaufschlagt. Der Abscheidegrad wird für 0,4 µm bestimmt.
Zur Modellierung haben wir vereinfachend unterschieden zwischen Schwebstofffiltern mit polydispersen Fasern
und Vorfiltern mit monodispersen Fasern
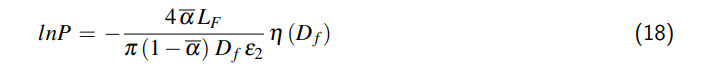
wobei α = mittlerer Faservolumenanteil (Packungsdichte), LF = Dicke des Filtermediums, Df = Faserdurchmesser, D*f = wirksamer Faserdurchmesser,{ Df }= mittlerer Faserdurchmesser,{ D2f } = mittleres Faserdurchmesserquadrat, ε1 bzw. ε2 = Korrekturfaktoren zur Anpassung an experimentelle Wert,
η = Einzelfaser-Abscheidegrad. Die verwendeten Strukturdaten sind in Tab. (2) zusammengestellt. Der Einzelfaser-Abscheidegrad η wurde für den Fall der polydispersen Mikroglasfasem der Schwebstoff-Filter nach der Theorie von Fuchs-Stechkina-Kirsch [8,9] berechnet. Für den Fall der monodispersen Vorfilter haben wir die vereinfachte Theorie nach Lee [10] verwendet in Kombination mit der Gleitreibungskorrektur von Rubow [11].
6 Berechnungsgleichung
Die Herleitung der Berechnungsgleichung kann hier nur skizziert werden, nähere Einzelheiten siehe Aufsatz von Ziemer und Schenderlein [5]. Man stellt zunächst eine raumseitige Bilanz der Fraktionsteilchenströme auf:
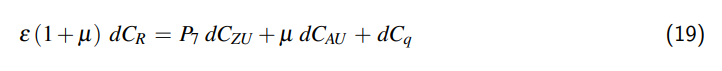
Auf der linken Seite steht bereits die gesuchte Größe dCR, auf der rechten Seite die noch unbekannte Größe dCZU , die aus einer anlagenseitigen Bilanz ermittelt wird:
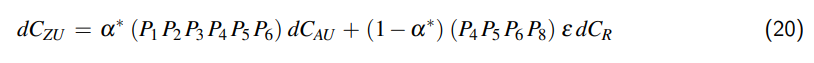
Man setzt nun Gl. (20) in Gl. (19) ein und stellt nach der gesuchten Größe dCR um:
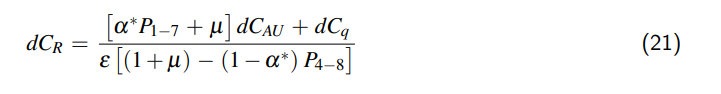
Um einen Vergleich mit den Reinheitsklassen nach ISO 14644-1 oder EG-Leitfaden, Annex 1 durchführen zu können, die ja bekanntlich kumulative PGVTen sind, muss Gl. (21) integriert werden:
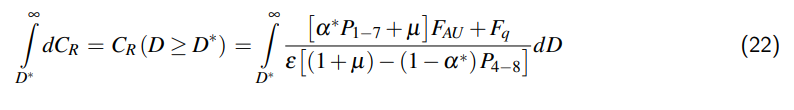
Die Größen bedeuten: α* = α/(1−λ) = der um den Leckluftanteil reduzierte Außenluftanteil mit α = V˙AU /V˙ ZU = Außenluftanteil und λ = V˙ LE/V˙ ZU = Leckluftanteil, µ = V˙ INF/V˙ ZU = Migrationskoeffizient, d.h. der Außenluftanteil, der durch die Fassadenfugen aufgrund der Windauflastung in den Reinraum eindringt (Infiltration), ε = Lüftungseffektivität, P1−7 = Produkt der fraktionellen Penetrationen der Filter (l) bis (7) und P4−8 = desgleichen für die Filter (4) bis (8). Die Penetrationen werden nach den in Abschnitt (5) beschriebenen Verfahren berechnet.
7 Ergebnisse
In Abb. (6) sind typische PGVTen dargestellt, wie sie sich im Reinraum unter Arbeitsbedingung (in operation) und dem Einfluß verschieden starker Außenluftinfiltration einstellen würden. Die gewählte Filterbestückung bestand aus einem Vorfilter P4 = F7 und einem Nachfilter P6 = F9 im Umluftgerät (P1 = P2 = P3 = P5 = 1 gesetzt). Ferner wurde ein endständiges Schwebstoffilter P7 = H 14 verwendet und auf ein raumseitiges Abluftfilter verzichtet (P8 = 1). Bei der unteren Kurve (µ = 0%, idealdichte Fassade) erkennt man am rechten Teil die Kontamination durch die im Reinraum anwesenden Personen. Sie ist wegen der geringen Besetzungsdichte (1 Person/50 m2 ) gering, für D > 0,4 µm ca. C = 2 500 1/m3 bzw. 70 1/ft3 .
Unterhalb von 0,4 µm wird die Personenkontamination völlig überdeckt von den Außenluftteilchen, die alle o.g. Filterstufen durchdringen. Zwischen D = 0,4 … 0,15 µm erfolgt ein steiler Anstieg (ca. 40fach) der Anzahlkonzentration bis ca. 1 · 104 m3 bzw. 285 1/ft3 . Diese Partikelgröße liegt zwar außerhalb der in der Pharmazie üblichen Betrachtungsgrenzen, die Teilchen stellen aber doch eine gewisse chemische Kontamination dar (z. B. ultrafeine Rußteilchen). Sobald eine gewisse Fassadenleckage auftritt, steigt der Aerosolgehalt im Reinraum an. Bei µ = 0,5% wird dann bereits die Grenzkurve der Reinheitsklasse B überschritten. Wenn man die spezifische Leckluftrate γ = V˙ INF/AW berechnet, erhält man:
Wenn man auf Dauer sicher die Reinheitsklasse C (in operation) nach EG-Leitfaden, Annex 1 [15] einhalten will, empfiehlt es sich
als spezifische Fassadenleckage einzuhalten. Die wenigen bisher vorliegenden experimentellen Bestimmungen an Fassaden zeigen, dass dieser Wert auch eingehalten werden kann.
δ μ ≅
Literatur
1
Sockel, H.: Aerodynamik der Bauwerke, Friedr. Vieweg & Sohn, Braunschweig, 1984
2
Fuchs, N. A.: The Mechanics of Aerosols, Pergamon Press, Oxford, 1964
3
Friedlander, S. K.: Smoke, Dust and Haze- Fundamentals of Aerosol Behavior, John Wiley & Sons, New York, 1977
4
Hinds, W. C.: Aerosol Technology- Properties, Behavior and Measurement of Airborne Particles, John Wiley & Sons, New York, 1982
5
Ziemer, W. and Schenderlein, S.: Particle Size Distribution in Pharmaceutical Cleanrooms, Proceedings 12th Int. Symp. on Contamination Control, Oct. 10 to 14, 1994, Yokohama, Japan, pp. 79-86
6
Junge, C. E.: Gesetzmäßigkeiten in der Größenverteilung atmosphärischer Aerosole über dem Kontinent, Ber. d. Deutschen Wetterdienstes in der US-Zone, Nr. 35, S. 261- 277, Bad Kissingen, 1952
7
Minamino, O. and Fujii, S.: Generation of Dust from Garments Worn in the Cleanroom, Proceedings 8th Int. Symp. on Contamination Control, Sept. 9 to 12, 1986, Milan, Italy, pp. 178 181
8
Fuchs, N.A.; Kirsch, A. A. and Stechkina, I. B.: A Contribution on the Theory of Fibrous Aerosol Filters, Faraday Symposia of the Chemical Society, No. 7: Symposium on Fogs and Smokes, 28- 30 March, 1973, pp. 143- 156
9
Kirsch, A. A. and Stechkina, I. B.: The Theory of Aerosol Filtration with Fibrous Filters, Chapter 4, pp.:165-256, in Fundamantals of Aerosol Science, ed. D. T. Shaw, John Wiley & Sons, New York, 1978
10
Lee, K. W.: Filtration of Submicron Aerosols by Fibrous Filters, Ph. D., 1977, University of Minnesota, Microfilm No. 78- 2688
11
Rubow, K. L.: Submicron Aerosol Filtration Characteristics of Membrane Filters, Ph. D., 1981, University of Minnesota, Microfilm No. 82- 11537
12
Ziemer, W. und Förster, B.: Partikeltransport durch undichte Fassaden, Reine Technologien Aktuelle Fragen der Reinraumtechnik, VDI Berichte 1238, pp. 27-39, Karlsruhe, 1996
13
DIN EN 779:2003-05: Partikel-Luftfilter für die allgemeine Raumlufttechnik- Bestimmung der Filterleistung, 70 Seiten
14
DIN EN 1822-1:1998-07: Schwebstoffilter (HEPA und ULPA)- Teil 1: Klassifikation, Leistungsprüfung, Kennzeichnung, 10 Seiten
15
EU Guidelines to Good Manufacturing Practice, Volume 4, Annex 1: Manufacture of Sterile Medicinal Products, European Commission, 14 February 2008, Brüssel
1 Einleitung
Reindampf wird in der Pharmazeutischen Industrie vorwiegend zur Sterilisation verwendet. Dies beruht darauf, dass Wasser sich hervorragend als Energieüberträger eignet. Bei der Verdampfung wird eine große Menge Energie benötigt, die bei der Kondensation kurzfristig abgegeben werden kann. In der Dampftabelle sind die Enthalpiewerte für trockenen, gesättigten Wasserdampf dargestellt.
Erste Voraussetzung für das Speisewasser ist, dass es Trinkwasserqualität besitzt. Weitere Vorgaben an das Speisewasser werden von dem Hersteller definiert.
Diese Vorgaben werden von gereinigtem Wasser (PW- purifed water) eingehalten, so dass in der Praxis das Reindampfsystem aus einem bestehenden PW-System gespeist wird. Das Speisewasser sollte entgast werden, bevor es in den Verdampfer gelangt. Dies ist notwendig, um die Menge der nichtkondsierbaren Gase (NKG) zu reduzieren und bei der Leitfähigkeitsmessung aufgrund des gelösten CO2 keine Störgröße zu bekommen. Gerade bei einer online Leitfähigkeitsmessung, bei der die Leitfähigkeit nur nach der Stufe 1 entsprechend der Pharmakopöe bewertet wird, kann es zu Überschreitungen aufgrund des gelösten CO2 kommen. Als Abhilfe kommt entweder eine Membranentgasung oder ein Entgasungsbehälter zum Einsatz.
Zur Erzeugung des Dampfes kommen dann das Naturumlaufverfahren, das Fallstromverfahren oder die Reindampferzeuger mit externen Wärmetauschern zum Einsatz.
Die Erzeuger sind zusätzlich mit Tröpfchenabscheider versehen, um ein Mitreißen von Kondensat und der darin gelösten Verunreinigungen, wie beispielsweise Endotoxine, zu verhindern.
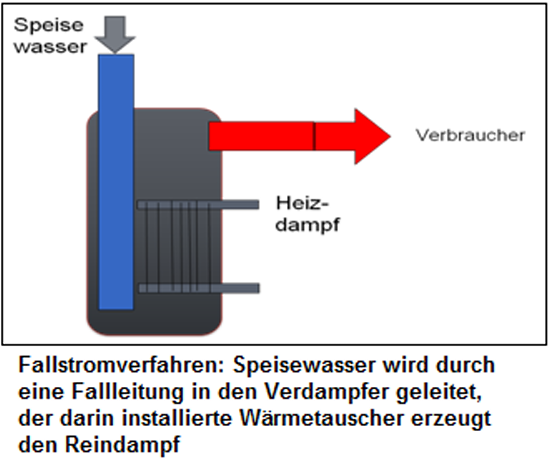
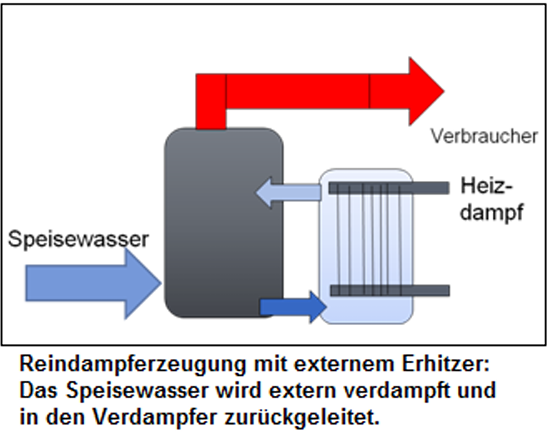
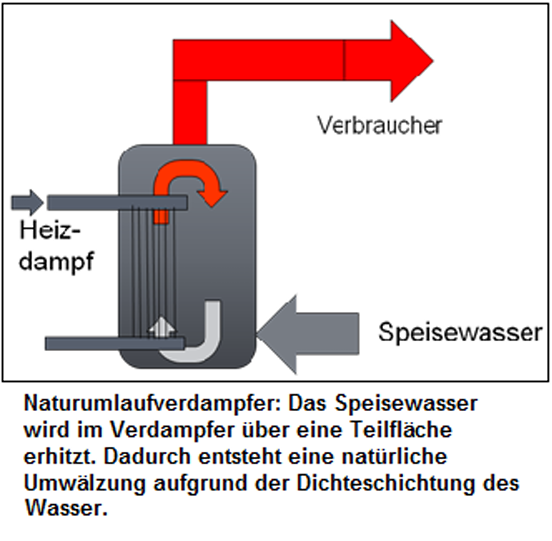
Betrieb des Reindampferzeugers
Um das Reindampfsystem betreiben zu können, muss die Temperatur und der Druck gemessen werden. Diese Parameter werden zur Steuerung der Anlage benötigt. Wichtig hierbei ist, dass Reindampf durch Verdampfung bei mindestens 100 C erzeugt werden muss und keine Zusätze enthalten darf. Zuzüglich zu diesen beiden Parametern müssen weitere qualitätsrelevante Parameter überprüft werden, die in den entsprechenden Regularien festgelegt sind. Zum einen sind Anforderungen an das Kondensat des Reindampfes in der Monografie Pure Steam der USP [7] gefordert, diese entsprechen den Anforderungen von Water for Injection (WfI). Das heißt, dass der Endotoxingehalt nicht größer als 0,25 EU / mL sein darf und die Leitfähigkeit und der TOC-Gehalt müssen den Anforderungen der Kapitel <643> und <645> der USP [7] entsprechen. Darüber hinaus sind in der Norm DIN EN 285 [1] und in der Norm DIN 59850 [2] Vorgaben über die Reindampfqualität festgelegt. In diesen beiden Normen werden zusätzlich zur Überprüfung des Kondensats noch Grenzwerte für die Eigenschaften des Dampfes angeben. Die zu untersuchen den Parameter sind: Überhitzung, Trockenheit und nicht kondensierbare Gase. Die Prüfungen können entweder mit der in der DIN EN 285 [1] beschriebenen offine-Methode, oder jedem vergleichbarem Messsystem durchgeführt werden. Bei den verfügbaren, alternativen Systemen besteht die Möglichkeit diese drei Parameter, die im Folgenden genauer vorgestellt werden, online zu bestimmen.
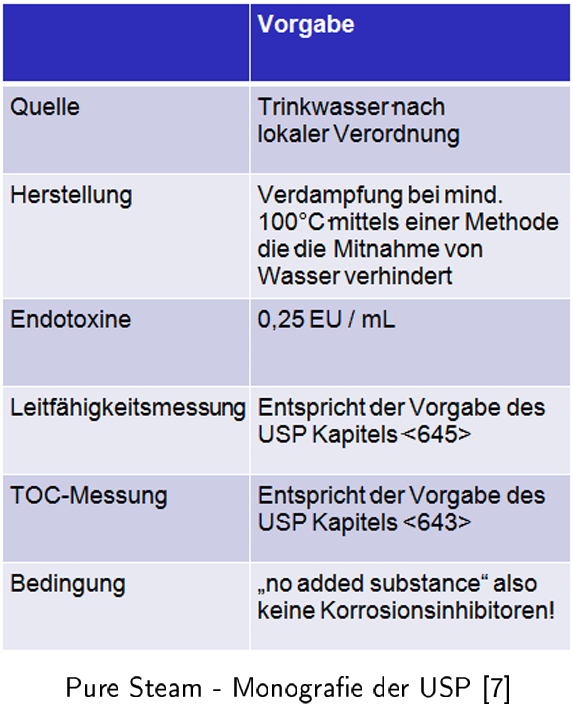
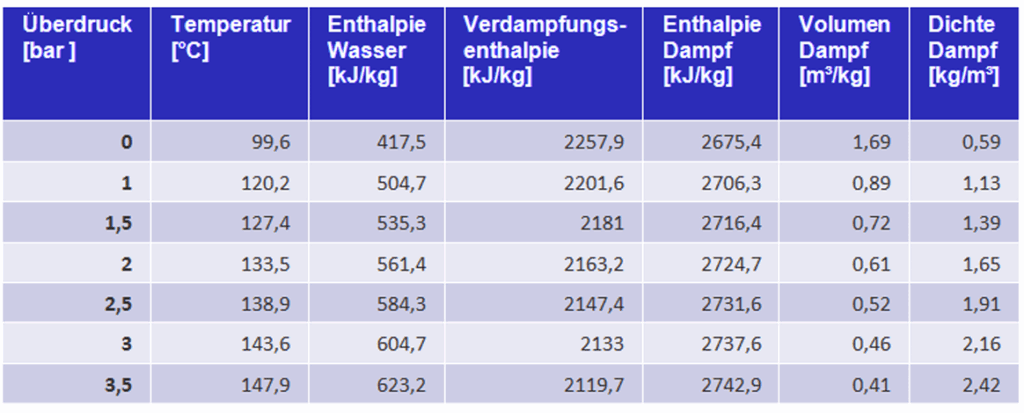
Nichtkondensierbare Gase
Messungen gemäß der DIN EN 285
Die nichtkondensierbaren Gase (NKG) verhindern einen direkten Kontakt des Dampfes mit dem Sterilisiergut und machen eine vollständige Sterilisation unmöglich. Während des Prozesses soll der Dampf kondensieren und mit der dabei übertragenden Enthalpie mögliche vorhandene Biomasse zerstören. NKGs wirken wie ein Isolator und verhindern die direkte Energieübertragung durch Ausbildung von Gasschichten oder Taschen. NKGs werden durch das Speisewasser eingebracht, speziell wenn keine Entgasung vorgeschaltet ist. Dies wird gerade dann begünstigt, wenn sich das Speisewasser über einen längeren Zeitraum in einem Lager und Verteilsystem befindet, das zur Atmosphäre geöffnet ist. Durch die permanente Umwälzung und den Einsatz von Sprühkugeln kann sich Luft, insbesondere CO2 , im Speisewasser lösen. In der unten abgebildeten Tabelle sind die potenziell auftreten den NKGs zusammengefasst. Gemessen werden die NKGs am höchsten Punkt des Verteilsystems, da sich an dieser Stelle Gastaschen vermehrt ausbilden können. Der Dampf soll maximal einen Volumenanteil von 3,5 % NKGs im Kondensat haben.
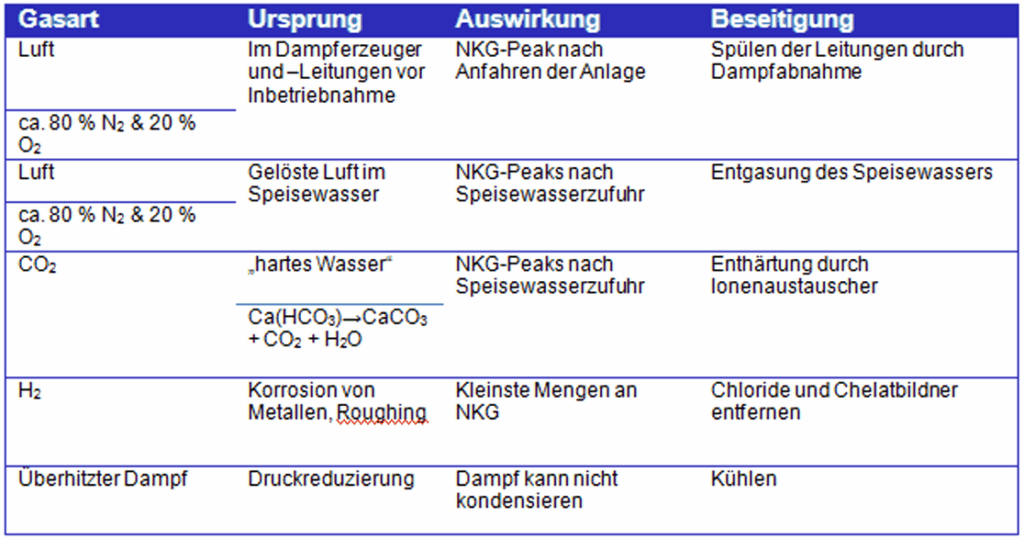
Trockenheit
Messungen gemäß der DIN EN 285
Dampf mit einem hohen Anteil an Kondensat (Nassdampf) reduziert die übertragene Energie während des Sterilisationsprozesses und führt nebenbei zu einer unerwünschten hohen Befeuchtung des Sterilgutes. Konstruktiv durch die Tröpfchenabscheider bedingt, ist der Dampf direkt nach dem Erzeuger trocken und gesättigt. Bei Fehlfunktionen dieser Abscheider kann durch eine unzureichen de Abscheidung Kondensat mitgerissen werden. Dies führt nicht nur zu Nassdampfbedingungen, sondern birgt auch das Risiko, dass Endotoxine mitgeführt werden. Weitere Nassdampfbildung kommt durch Kondensation im Verteilsystem zu stande. Daher haben die Dimensionierung, die Isolierung und die Entwässerung des Verteilsystems einen großen Einfluss auf die Trockenheit.Die Messung sollte nahe am Verbraucher durchgeführt werden. Als Akzeptanzkriterium gibt die DIN EN 285 [1] einen Trockenheitswert von 0,95 für Metallbeladungen und 0,9 für die restlichen Beladungen an.

Überhitzung
Überhitzter Dampf kann beim Sterilisationsvorgang nicht kondensieren, sondern muss sich erst bis auf den Siedepunkt abkühlen. Vorher kann keine Kondensationsenthalpie an das Sterilisiergut abgegeben werden und der überhitze Dampf verhält sich wie heiße Luft. Für eine erfolgreiche Sterilisation wären entweder eine längere Verweilzeit oder höhere Temperaturen nötig. Überhitzter Dampf kann aufgrund von großem Druckabfall im Verteilsystem oder im Sterilisator entstehen. Bei einer Druckerniedrigung bleibt die Gesamtenthalpie gleich. Diese Energieerhaltung führt erst zu einer kompletten Verdampfung von möglichem Kondensat und, nachdem Sattdampfbedingungen erreicht sind, zu einem Anstieg der Temperatur. Die Überhitzung von frei in die Atmosphäre ausströmendem Dampf darf maximal 25 K betragen.

Verteilnetz
Im Reindampfverteilnetz liegt, bedingt durch das kompressible Medium Reindampf, eine Expansionsströmung vor. Dabei nimmt der Druck aufgrund von Reibungsverlusten im Rohrleitungsverlauf ab. Eine Übersicht über Druckverluste im idealen System ist in der nebenstehenden Tabelle wiedergegeben. Die Dampfgeschwindigkeiten liegen bei 25-40 m/s.
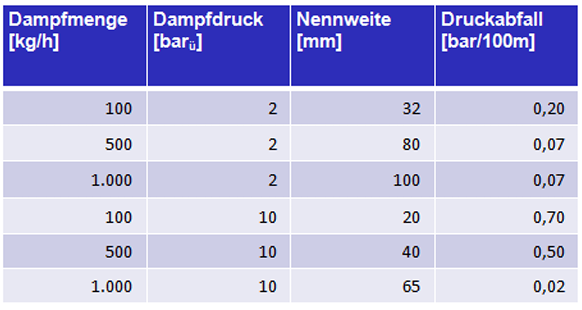
Die Rohrleitung dehnt sich bei der Inbetriebnahme aufgrund des Aufheizens aus. Diesem Effekt muss bei der Planung Rechnung getragen werden, da es sonst zu Spannungen im Verteilnetz kommen kann. Die Halterungen eines Reindampfverteilnetzes müssen bei geraden Strecken eine axiale Verschiebung zulassen. In längeren Leitungsabschnitten müssen zusätzlich Dehnungsausgleicher installiert werden. Diese müssen so konstruiert sein, dass sich keine Tiefpunkte bilden. Festpunkte sind so zu montieren, dass die Rohrbewegung keinen Schaden anrichten kann. Um Energieverluste, Kondensatbildung und eine Gefährdung von Mitarbeitern zu minimieren, muss das Verteilnetz isoliert werden. Die Isolierung erfolgt im Technikbereich aluminiumkaschiert und im Reinraumbereich als Rohr-in-Rohr-System.
Kondensat
Da es sich bei Dampfleitungen nicht um adiabatische Systeme handelt, wird immer ein Teil des Dampfes kondensieren und muss aus dem Verteilnetz abgeführt werden. Gerade beim Anfahren wird eine groÿe Men ge von Dampf in dem noch nicht erhitzten Verteilnetz kondensieren. Das Verteilnetz muss so konstruiert sein, dass das Kondensat komplett ablaufen kann. Die Abscheidung erfolgt über Kondensatableiter, von denen unterschiedliche Konstruktionen auf dem Markt erhältlich sind. Die Kondensatabscheider müssen an allen Tiefpunkten des Verteilnetzes installiert werden. Das Gefälle der Rohrleitung zu den Tiefpunkten sollte nicht kleiner als 1% sein. Das Kondensatnetz, das an das Reindampfsystem angeschlossen ist, sollte drucklos ausgeführt werden.
Entlüften
Beim abgeschalteten Reindampfsystem befindet sich hauptsächlich Luft im gesamten Verteilnetz. Beim Anfahren wird diese aufgrund des Druckanstieges komprimiert. Der Anteil der nichtkondensierbaren Gase darf wie in der DIN EN 285 [1] beschrieben einen Maximalanteil von 3,5 % haben. Über einen Entlüfter kann die im System befindliche Luft ausgetragen werden. Eine Vermischung von Dampf und Luft lässt sich durch eine zeitnahe Entlüftung nach dem Anfahren verhindern.
Literatur
1
DIN EN285:Sterilisation- Dampf-Sterilisatoren- Groß-Sterilisatoren, Deutsche Fassung EN 285:2006 + A2:2009, August 2009
2
DIN-Reihe 58950: Sterilisation- Dampf-Sterilisatoren für pharmazeutische Sterilisiergüter, 2011
3
DIN EN ISO 17665-1: Sterilisation von Produkten für die Gesundheitsfürsorge- Feuchte Hitze Teil 1: Anforderungen an die Entwicklung, Validierung und Lenkung der Anwendung eines Sterilisationsverfahrens für Medizinprodukte, Deutsche Fassung EN ISO 17665-1:2006, November 2006
4
ISO 13824: Bases for design of structures- General principles on risk assessment of systems involving structures, 42 Seiten, November 2009
5
EU Guidelines to Good Manufacturing Practice, Volume 4, Annex 1: Manufacture of Sterile Medicinal Products, European Commission, 25 November 2008 (rev.), Brüssel
6
ISPE Baseline R Guide, Volume 4: Water and Steam Systems (Second Edition), Department of health and human services, 01 Novomber 2011, Florida
7
The United States Pharmacopeia, 35th Edition and The National Formulary, 30th Edition, Rockville 2011
8
WHO Technical Report Series, No. 929, Annex 3: Good Manufacture Practices: water for pharma ceutical use, World Health Organization, 2005
Einleitung
Das mikrobiologische Monitoring stellt sicher, dass pharmazeutische Zubereitungen unter kontrollierten Bedingungen hergestellt werden. Da bei werden Abweichungen vom validierten Grundzustand festgestellt. Treten dabei Abweichungen auf (Out-of-Specication Resultate, OOS oder Out-of-Trend Resultate, OOT), ist man in der Lage korrigierende Maßnahmen ein zuleiten. Das mikrobiologischen Monitoring um fasst dabei die Überwachung der Luft, des ein gesetzten Prozesswassers, der Oberflächen und des Personals.
Monitoring-Programm Umgebung
Für die Überwachung der Reinräume sollte ein umfassendes Monitoring-Konzept erarbeitet werden. Das Monitoring-Programm muss in SOPs schriftlich festgelegt sein. Darin sollten folgende Angaben zu den Probenahmen enthalten sein: Messstellen, Frequenz, Zeitpunkt, Verantwortlichkeiten, Dokumentation, Equipment und Techniken. Weiterhin müssen die Warn- und Aktionsgrenzen festgelegt sein. In der SOP sollte auch der formale und zeitliche Ablauf, wenn es zu einer Überschreitung der Warn- bzw. Aktionsgrenzen kommt, festgelegt sein
Warn- und Aktionsgrenzen
Um die Qualität beurteilen zu können, müssen die Anforderungen an die Reinräume beschrieben sein. Diese Anforderungen werden als Warn bzw. Aktionsgrenzen festgelegt. Bei den Aktionsgrenzen kann auf die in den Regelwerken (z.B. USP, Annex 1 (Sterilfertigung) EU-GMP Leitfaden, FDA, Guidance for Industry) beschriebenen Grenzen zurückgegriffen werden.

Wenn die Aktionsgrenze selber festgelegt werden muss, kann man eine der folgenden Regeln anwenden:
- liegt z.B. außerhalb des statistisch wahrscheinlichen Bereichs
- oder (z.B. bei sehr kleinen Messgrößen/ seltenen Einzelfunden)
- überschreitet Basislevel um 50% oder 100% [8]
Bei den Warngrenzen sollte man dagegen auf historische Daten oder auf Ergebnisse während der Qualifizierungsphase zurückgreifen. Die Warngrenze entspricht dabei der Überschreitung des Bereichs, in dem die Messdaten laut Datenstatistik am häufigsten lagen. Eine Berechnung kann dann zum Beispiel anhand des Vorschlags in der DIN/EN 1632 (Entwurf, nicht realisiert) erfolgen. Wenn die Warngrenze nicht statistisch ermittelt werden kann, wird auch oft 1/2 Aktionsgrenze als Warngrenze vorläufig festgelegt. Eine Überschreitung der Warngrenze zieht nicht zwangsläufig eine vollständige Untersuchung nach sich. Hierbei wird lediglich eine potentielle Drift von den Normalbedingungen festgestellt. Aktionsgrenzüberschreitungen müssen dagegen immer auf deren Ursache untersucht und dokumentiert werden.

Umgang mit Abweichungen
Treten Abweichungen auf, so befindet sich die Produktionsumgebung nicht mehr unter Kontrolle. Die Überschreitung der erforderlichen Umgebungsbedingungen muss zeitnah untersucht werden und innerhalb von 30 Tagen abgeschlossen sein. Dabei soll die Ursache gefunden werden, um entsprechende korrigierende Maßnahmen (Corrective Action) einleiten zu können. Weiterhin sollten vorbeugen de Maßnahmen (Preventive Action) veranlasst werden, da mit diese Abweichungen nicht noch einmal auftreten. Man spricht auch von einem CAPA-Plan.
Datenreview
Die vorhandenen Daten aus dem Monitoring Programm sollten regelmäßig zusammengefasst und bewertet werden. Die zeitlichen Abstände können individuell festgelegt sein und hängen von der Anzahl der Probenahmen und der Kritikalität (Rolle im Prozess und Monitoring-Ergebnisse über Warngrenze) der untersuchten Reinräume ab. Der Daten review kann auch zu einer Neubewertung der Warngrenze herangezogen werden.
Erkennen von Trends
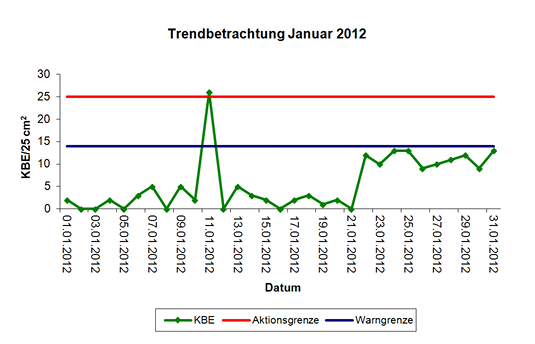
Im Rahmen des Hygienemonitorings sollten Trendanalysen durchgeführt werden. Diese Forderung ist mittlerweile auch in die Anforderungen der WHO eingegangen [9]. Dabei können sich abzeichnende Abweichungen vom validierten Zustand, z.B. beim gehäuften Auftreten von Warngrenzen, frühzeitig erkannt und korrigierende Maßnahmen eingeleitet werden (OOT-Out of Trend- Ereignisse). Auch hier findet man von amtlicher Seite in der einschlägigen Literatur Hinweise:
„When data are compiled and analyzed, any trends should be evaluated by trained personnel. While it is important to review environmental results based on recommended and specified frequency, it is also critical to review results over extended periods to determine whether trends are present. Trends can be visualized through the construction of statistical control charts that include alert and action levels. The microbial control of controlled environments can be assessed, in part, based on these trend data.“ [4]

„Levels of detection of microbial contamination should be established for the purpose of setting alert and action limits and for monitoring the trends in environmental cleanliness in the facility. „[9]
„The quality control unit should provide routine oversight of near-term (e.g., daily, weekly, monthly, quarterly) and long-term trends in environmental and personnel monitoring data. [5]
„Daten, die aufgrund einer einzigen Probe ermittelt wurden, sind oft nicht signifikant. Darüber hinaus können mikrobiologische Überwachungstechniken ernsthafte Mängel haben, die eine breite Streuung verursachen. Eine grafische Darstellung der gesammelten Daten über eine bestimmte Zeitspanne kann deshalb zur Unterscheidung der Streuungen in der Probenahme von tatsächlichen Trends nützlich sein oder anzeigen, dass eine signifikante Änderung eingetreten ist, obwohl die Schätzwerte innerhalb der festgelegten Grenzwerte liegen. [7]

Um das Erkennen von Trends zu erleichtern, ist es oft zweckmäßig, die Daten als gleitende Mittelwerte z.B. aus 5-10 aufeinander folgenden Einzelwerten darzustellen.
Außerdem sollte das Trending auch in Bezug auf die isolierten Keimarten erfolgen. So können, z.B. beim gehäuften Auftreten von Sporenbildnern oder Schimmelpilzen, rechtzeitig korrigierende Maßnahmen eingeleitet werden.
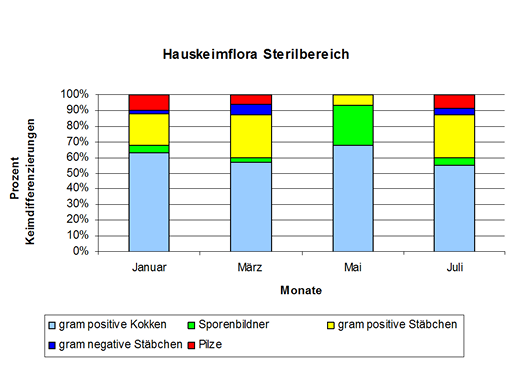
Literatur
1
EU Guidelines to Good Manufacturing Practice, Volume 4, Annex 1: Manufacture of Sterile Medicinal Products, European Commission, November 2008, Brüssel
2
ISO 13408-1:2008-06: Aseptic processing of health care products- Part 1: General requirements, 36 Seiten
3
ISO 14698-1:2004-04: Reinräume und zugehörige Reinraumbereiche- Biokontaminationskontrolle- Teil 1: Allgemeine Grundlagen, 38 Seiten
4
USP 29 <1116>: Microbiological evaluation of clean rooms and other controlled environments
5
FDA: Guidance for Industry- Sterile Drug Products Produced by Aseptic Processing- Current Good Manufacturing Practice, 2004
6
Parental Drug Association (PDA): Technical Report No. 13
7
Wallhäusers Praxis der Sterilisation, Thieme Verlag, 2008
8
WHO Technical Report Series, No. 961, Annex 6: WHO good manufacturing practices for sterile pharmaceutical products, World Health Organization, 2011
Abstract
Die Reinigung von Equipment ist eine Disziplin, die ein hohes Maß an chemischem, toxikologischem, pharmakologischem und verfahrenstechnischem Know-how erfordert. Nur wenn alle fachlichen Disziplinen zusammenarbeiten, lässt sich ein möglichst wirtschaftliches und dennoch zuverlässiges Verfahren für multipurpose-Anlagen etablieren und validieren. In diesem Artikel sollen chemische, toxikologische und pharmakologische Aspekte zur Planung und Auslegung des Reinigungsverfahrens und verfahrenstechnische Bewertung von Bracketing und Probenahme diskutiert werden.
1 Einleitung und Historie
Der Grundstein für die Notwendigkeit der Reinigungsvalidierung wurde mit der Einführung der Penicilline gelegt. Im FDA „Guide to In spections- Validiation of Cleaning Processes“ 7/93 [5] heißt es, dass die meisten Produktrückrufe aufgrund von Kreuzkontamination durch Penicilline hervorgerufen wurden. Im Hinblick auf die Reinigung waren auch die mangelnde Hygiene und fehlende Staub Kontrolle ein weiterer Grund für die Behörden, regulatorische Bestimmungen festzulegen. Der FDA Guide stammt aus dem Jahr 1993 und enthält alle wesentlichen Grundzüge der Reinigungsvalidierung. Der Guide bezieht sich auf die Herstellungsprozesse der chemischen und der biotechnologischen Pharmaindustrie. Die Gründe zur Entstehung dieses Guides liegen in zuvor aufgetretenen Skandalen wie z.B. Pestizidrückständen in Arzneimitteln und mangelhafter Beweisführung für die Abwesenheit von Rückständen in einer multi-purpose Anlage in der auch Steroide hergestellt wurden.
Heute muss für jede Reinigungsvalidierung eine risikobasierte Betrachtung sämtlicher Stoffe, die in das nachfolgende Produkt gelangen können, erfolgen. Hierzu zählen Rückstände von Wirkstoffen, Reinigungsmitteln und möglichen Abbauprodukten. Um mögliche Kreuzkontaminationen im Folgeprodukt zu vermeiden, werden Akzeptanzkriterien definiert. Diese werden gemäß ICH Q9 [9] risikobasiert für jeden einzelnen Wirkstoff einer Mehrzweckanlage festgelegt. Akzeptanzkriterien für mikrobiologische Grenzwerte wer den nicht genannt, jedoch werden die mikrobiologischen Aspekte in den einschlägigen Regelwerken und Empfehlungen wie dem FDA Guide [5], EU-GMP-Leitfaden, Annex 15 [4] und PIC/S [11] erwähnt.
2 Grenzwerte
für Rückstände von Produkten, Reinigungsmitteln und möglichen Abbauprodukten
Mit dem 2015 revidierten Annex 15 EU GMP Leitfaden wurde ein neuer Ansatz für die Betrachtung von möglichen Rückständen veröffentlicht. Mit den Neuerungen sind die bisherigen Akzeptanzkriterien für Produktrück stände, 1/1000 Dosiskriterium und 10-ppm Mengenkriterium nicht mehr allein zu verwenden. Vielmehr sollte das angewandte Kriterium aufgrund einer Risikobewertung der Kontaminanten erfolgen, die neben toxikologischen Betrachtungen auch die Bewertung der pharmakologischen und physikalisch-chemischen Eigenschaften (z.B. Löslichkeiten) beinhalten. Die bisherigen Grenzwerte sollen durch den wissenschaftsbasierten Grenzwert für die tägliche Exposition PDE (Permitted Daily Exposure) oder einen TTC (Threshold of Toxicological Concern)-Wert abgelöst werden. Dieser ist für jeden einzelnen Wirkstoff und jedes Reinigungsmittel zu ermitteln. Für generische Wirkstoffe kann die Ermittlung der Werte über die Beauftragung der Erstellung eines entsprechenden Gutachtens erfolgen.
Stehen die Werte nicht zur Verfügung, z.B. bei Neuentwicklungen, besteht für Arzneimittel-Hersteller die Möglichkeit dass Mengen- oder Dosiskriterium in Verbindung mit dem OEL-Wert (Occupational Exposure Limit) anzuwenden. Hierzu muss der Hersteller nach weisen, dass das bisher verwendete Kriterium größer oder gleich dem jeweiligen OEL-Wert des Wirkstoffes, inklusive Betrachtung des Aufnahmeweges, ist. Der OEL-Wert stellt einen aus toxikologischen Daten hergeleiteten Grenzwert dar.
Gründe dieser Neuerungen sind zum einen, dass das Dosis- und Mengenkriterium willkürlich ohne risikobasierte Ansätze gewählt wurde und keine wissenschaftliche Betrachtungsweise bestand. Der unterschiedliche therapeutische Index von Arzneistoffen fand keine Berücksichtigung. Zudem ist die therapeutische Dosis proportional zum Rückstand, was bei hochdosierten Stoffen zu hohen Produktrück ständen führt. Außerdem müssen Langzeitintoxikationen und teratogene toxische Effekte in die Risikobetrachtung einbezogen werden.
2.1 PDE-Kriterium
PDE(Permitted Daily Exposure) beschreibt die Dosis eines Stoffes, bei der bei täglicher Aufnahme über den gesamten Lebenszeitraum kein negativer Effekt beobachtet wird. Das Akzeptanzkriterium stützt sich auf wissenschaftliche Daten wie klinische oder toxikologische Studien und enthält eine Risikobetrachtung. ADE (Acceptable Daily Exposure) stellt ein Synonym des Kriteriums dar. Die Berechnung der Dosis wird auf Basis von toxikologischen Studien nach der EMA “Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities” durch geführt [3].
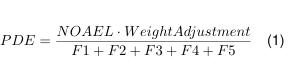
- PDE:Permitted Daily Expose [mg/Tag]
- NOAEL: Höchste Dosis, bei der kein nachteiliger kritischer Effekt zu beobachten ist (No Observed Adverse Effect Level) [mg/(Tag kg)] Weight Adjustment = Standard Körpergewicht 50 kg
- F1:Faktor zur Extrapolation zwischen den Spezies (2-12)
- F2: Faktor zur Unterscheidung zwischen den Spezies (10)
- F3: Faktor zur Berechnung von Kurz zeit/Langzeit Studien
- F4: Faktor bei schwerer Toxizität
- F5:Variabler Faktor wenn kein NOAEL bekannt, sondern PDE von LOEL (Lowest Observed Adverse Effect Level) abgeleitet wird.
Sollte der NOAEL nicht verfügbar sein, kann der LOAEL (Lowest Observed Adverse Effect Level) verwendet werden. Für produktspezifi sche Ausrüstung (Dedicated Equipment) be steht hinsichtlich von Wirkstoffrückständen kei ne generelle Validierungsplicht. Eine Bewertung bezüglich möglichen Abbauprodukten, Reinigungsmittelrückständen und mikrobiologischen Verunreinigungen muss jedoch erfolgen.
Anhand einer Risikobewertung sollte abgewogen werden, ob eine Mehrzweck oder produktspezifischen Anlage zur Herstellung der Arzneimittel verwendet werden kann. Bei der Ermittlung der PDE ist zu berücksichtigen, dass die Route der Administration des Folgeprodukts bekannt sein muss und in die Berechnung des PDEs einbezogen werden müssen. Bei einer subkutanen Anwendung kann z.B. bei vielen Lokalanästhetika eine höhere PDE toleriert werden, als für eine intravenöse Anwendung.
Die Reinigung der Anlagen nach der Benutzung erfordert oft den Zusatz von Reinigungssubstanzen. Auch diese können die Gesundheit der Patienten beeinträchtigen indem sie eine eigene toxische Wirkung entfalten oder die Wirksamkeit des Folgearzneimittels beeinträchtigen. Daher muss sowohl für Wirk- und Hilfsstoffe, als auch für die Reinigungsmittel ein Grenzwert ermittelt werden. Wie auch bei den Wirkstoffen muss dieser Grenzwert erreichbar und messbar sein. Bei einer Reinigungsroutine, die aus mehreren Schritten besteht, sollte man die Analytik auf die Last-to-rinse Substanz beziehen. Auf jeden Fall muss die Zusammensetzung des Reinigungsmittels bekannt sein und der Lieferant eine Langzeitgarantie auf die Rezeptur geben. Als Reinigungsmittel kommen üblicherweise Tenside in Frage, aber auch eine Abfolge von Säuren, Laugen und Komplexierungsmitteln ist üblich.
Wenn die PDE für die geplante Folgeanwendung ermittelt wurde, muss die maximal zulässige Rückstandsmenge MZR (=MACO) für alle Rückstände (Produkte, Abbauprodukte, Reinigungsmittel) berechnet werden.
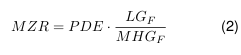
- MZR: Maximal zulässiger (Reinigungsmittel)Rückstand
- LGF: Chargengröße des Folgeproduktes (worst case)
- MHDF: Maximale humantherapeutische (Tages)dosis des Folgeproduktes = Einnahmehäufigkeit x Masse Darreichungsform
Bekannte chemische Strukturen mit unbekannten Toxizitäten können nach Allhenn und Anhalt [1] mit dem TTC-Konzept (Threshold of Toxicological Concern) beurteilt werden. (6) Genotoxische Stoffe oder Stoffe mit sensibilisierendem Potential können nicht über den TTC Wert definiert werden. Hierfür muss der PDE Wert oder die „Limits of Genotoxic Impurities“ [1] herangezogen werden.
2.2 GRAS-Status
Bei Substanzen, die GRAS-Status (generally regarded as safe) gemäß CFR 21 Part 184 [12] haben und keine bekannte PDE vorliegt, kann analog zu ICH Q3C das PDE für Restlösemittel mit niedrigem toxischem Potential PDE = 50 mg zugrunde gelegt werden [10].
2.3 10-ppm-Kriterium
Wenn die Toxizität der Stoffe sehr gering ist oder unbedenkliche Stoffe eingesetzt werden, empfiehlt es sich, dennoch einen „best practise“ Grenzwert festzulegen. Hier empfiehlt es sich das 10 ppm Kriterium als technisch machbaren Grenzwert zu verwenden. Maximal 10 ppm des Vorgängerproduktes dürfen in das Nachfolgeprodukt verschleppt werden. Damit ergibt sich für die Belastung der Folgecharge ein maximaler Wert an Rückstand mmax:
- mmax: Akzeptanzkriterium/ max. zulässiger Rückstand des Vorproduktes [mg]
- MCharge: minimale Chargengröße des Folgeprodukts [kg]
Das 10 ppm Kriterium hat seinen Ursprung in der Lebensmittelindustrie und berücksichtigt nicht die toxikologischen oder pharmakologischen Eigenschaften des Stoffes. Es ist jedoch weiterhin ein nützliches Kriterien, um bei toxikologisch unbedenklichen Stoffen ein Limit zu berechnen.
2.4 Das 1/1000 Dosiskriterium
Auch wenn das toxikologische Kriterium bei der Berechnung der maximalen Rückstandsmenge in Betracht zu ziehen ist, wird in Annex 15 gefordert, die pharmakologischen Eigenschaften und damit auch die Dosierung in die Risikobetrachtung einzubeziehen [4]. Daher sollte man weiterhin die übliche Dosierung des Folgeproduktes ermitteln und in die Diskussion des Grenzwertes einbeziehen.
In der Vergangenheit wurde die übliche bzw. auch die minimale Dosierung mit einem festen Risikofaktor der Grenzwertberechnung zugrunde gelegt. In der Tagesdosis des Folgeproduktes durfte nicht mehr als ein Tausendstel der niedrigsten therapeutischen Tagesdosis des Vorproduktes enthalten sein. Bezogen auf die kleinstmögliche Chargengröße so wie der Anlagenfläche erhält man die maximal zulässige Rückstandsmenge die von der produktberührenden Fläche auf die nächstfolgende Charge übergehen darf:
- MZR: Maximal zulässiger (Reinigungsmittel)Rückstand
- nTD: niedrigste therapeutische Dosis des Vorprodukts [mg/d]
- LGF: Chargengröße des Folgeproduktes (worst case)
- MHDF: Maximale humantherapeutische (Tages)dosis des Folgeproduktes = Einnahmehäufigkeit x Masse Darreichungs form
2.5 „Visually-Clean“- Kriterium
Die Anlagenoberfläche muss sichtbar sauber sein. Diese Forderung gilt generell bei der Reinigungsvalidierung. Allerdings ist eine Quantifizierung der Beobachtung „sauber“ schwierig. Der Versuch, das Kriterium für die visuelle Überprüfung zu quantifizieren wurde empirisch mittels „Spiking“-Studien (Verdünnungsstufen) für eine Gruppe von Substanzen ermittelt. Der Grenzwert, bei dem visuell die unter suchten Substanzen lagen, liegt etwa bei bei 4 µg / 100 cm² [6]. Da dieser Wert nur für wenige Produkte getestet wurde, kann er nicht auf alle Produkte übertragen werden. Ein Hersteller muss durch eigene „Spiking“-Studien belegen, ab welcher Konzentration ein Produkt gerade noch visuell erfasst werden kann. Dabei muss die Testoberflächeneigenschaft der Qualität der Anlagenoberfläche aus der Produktion entsprechen. Weiterhin stellen Produkte mit stark färbenden Eigenschaften ein Problem bei der Beurteilung der visuellen Sauberkeit dar. Es wird empfohlen die Unbedenklichkeit von Farbstoffen im Rahmen einer Risikoanalyse zu bewerten und gegeben falls die entsprechen den Produkte mit in die Reinigungsvalidierung einzubeziehen.
Das „Visually-Clean“ Kriterium berücksichtigt weder den Bezug zur Chargengröße noch zur Anlagenfläche. Deshalb ist dieses Kriterium immer im Zusammenhang mit dem PDE Kriterium und ggf. dem 10-ppm Kriterium zu betrachten und kann nicht als alleiniges Kriterium herangezogen werden.
2.6 Worst-case-Konzept
Nach Ermittlung der Grenzwerte wird die Kenntnis der PDE (und ggf. andere Grenz werte) dazu verwendet, produktabhängig einen Grenzwert zu berechnen. Man kann dann für das gesamte Produktspektrum ein worst-case Szenario aus Vorprodukt/Folgeprodukt bestimmen und diesen Grenzwert für die gesamte Reinigungsvalidierung betrachten. Dieser Grenzwert muss verifizierbar sein, d.h. ein validiertes Analysenverfahren muss diesen Grenz wert nachweisen können. Bei Herstellung neu er Produkte auf der Anlage muss die Gültigkeit des worst-case-Szenarios überprüft werden. Bei hochpotenten Wirkstoffen und Arzneimitteln wie Steroide, Antibiotika und Zytostatika ergibt die Risikobetrachtung, dass diese Produkte mit „dedicated equipment“ hergestellt werden müssen, weil z.B. ein ermittelter Grenzwert unterhalb der analytischen Nachweisgrenze liegt oder die Behörde dies bei bestimmten Produktgruppen fordert.
2.7 Umstellung von Dosiskriterium auf PDE
Wurde die Reinigungsvalidierung schon vor 2015 etabliert, mussten seitdem die in der Vergangenheit etablierten Grenzwerte hinterfragt und angepasst werden. Teilweise müssen bessere Reinigungsverfahren etabliert werden, teilweise ist der bisherige Grenzwert aber strenger. Eine Aufweitung von schon etablierten Grenzwerten ist nicht akzeptabel, da man eine technisch machbare Qualität der Reinigung beibehalten muss.
Eine toxikologische Betrachtung des Lokalanästhetikums „Bupivacain“ durch DPhE [8] er gab für die Reinigungsvaldierung, je nach Applikationsweg des Folgeprodukts, unterschiedliche Konsequenzen. Betrachtet wurden zwei Anlagen. Zur Ableitung von Maßnahmen bezüglich der Reinigungsvalidierung wird ein Bewertungsquotient aus dem alten 1/1000-Dosis Kriterium und dem neuen PDE benutzt.
Anlage A Die Folgeprodukte werden ausschließlich subcutan angewendet. Der Bewertungsquotient beträgt 0,2. Es sind keine Maß nahmen erforderlich.
Anlage B Die Folgeprodukte werden meist intravenös angewendet. Der Bewertungsquotient beträgt 3,9. Die Reinigung muss verbessert werden und die Reinigungsvalidierung muss erneut durchgeführt werden.
3 Mikrobiologische Grenzwerte
Während es bei der Betrachtung von Produkt und Reinigungsmittelrückständen auf die Vermeidung von Kreuzkontamination und Verschleppung durch Vorgängerprodukt und Reinigungsprozess ankommt, bezieht sich die Betrachtung der Keimzahl auf die Überwachung und die vorbeugenden Maßnahmen. Solche Maßnahmen sind zum Beispiel, dass das Equipment nach der Reinigung trocken gehalten werden muss. Die Zeiten zwischen Produktionsende und Reinigungsbeginn „dirty-hold time“ haben nicht nur Einfluss auf die Reinigung sondern auch auf die mikrobielle Belastung, was zum Beispiel in der Sterilfertigung eine besondere Rolle spielt. Ebenso müssen die Standzeiten zwischen Reinigungsende und Produktionsbeginn validiert werden. Diese bei den Gegebenheiten haben Einfluss auf das mikrobielle Wachstum. Die Grenzwerte für Keim zahlen an Oberflächen können hierbei aus den Anforderungen aus Annex 1 des EG-GMP Leitfadens [2] bei sterilen Arzneimitteln abgeleitet werden. Eine weitere Orientierung hinsichtlich der mikrobiologischen Reinheit von Arzneimitteln sind die Spezifikationen der jeweiligen Arzneibücher. Für Phytopharmaka gibt das europäische Arzneibuch Hinweise zur mikrobiellen Reinheit von Drogen in den verschiedenen Verarbeitungsstufen. Die mikrobiologichen Grenzwerte für die Reinigungsvalidierung können somit in diesem Fall aus den Produktanforderungen abgeleitet werden.
Eine Sonderstellung stellt hier die Sterilfertigung dar. Obwohl der Sterilisationsnachweis des Equipments durch die Validierung von Sterilisationsprozessen erbracht wird, kann den noch nicht sichergestellt werden, dass Pyrogene bzw. Endotoxine beseitigt wurden. Unter diesem Aspekt ist auch die Mikrobiologie im Rahmen der Reinigungsvalidierung und Standzeitvalidierung von Reinigungsverfahren in der Sterilfertigung nicht außer Acht zu lassen [5, 11].
4 Anlagendesign
Mit Hilfe einer Risikoanalyse soll der Einfluss von produkt-, anlagen- und prozessbezogenen Parametern auf das Reinigungsziel bewertet werden. Um den Aufwand der Reinigungsvalidierung zu reduzieren, kann durch eine Ähnlichkeitsbetrachtung hinsichtlich des Designs der Anlagen und Produkte mit vergleichbaren chemisch-physikalischen Eigenschaften eine Gruppierung, das sogenannte „Bracketing“, vorgenommen werden. Dadurch müssen ähnliche Produkte und Prozesse nicht einzeln validiert werden. Mögliche Kriterien zur Bildung von Gruppen sollen an einem Beispiel eines Rührbehälters dargestellt werden (siehe Tab. 1)
In diesem Beispiel wurde der Fokus auf Konstruktionsmerkmale gelegt, die sich signifikant auf die Reinigung auswirken. Damit konnte eine risikobasierte Rationale für die Zusammenfassung von Anlagen mit unterschiedlichen Konstruktionsmerkmalen dargelegt wer den. Im nächsten Schritt wird dann in jeder Gruppe der worst-case Behälter ermittelt und damit die Reinigungsvalidierung durchgeführt. Für ein erfolgreiches Reinigungsverfahren sind leicht zu reinigende Komponenten unumgänglich. Die reinigungsgerechte Gestaltung der Anlagen und Anlagenteile wird mit dem Hygienic Design beschrieben. Jede produktberührende Oberfläche muss mit dem Reinigungsmittel benetzbar und einfach zu trocknen sein. Bei der hygienegerechten Gestaltung der Anlage tragen auch der Werkstoff und die Oberfläche der Anlage eine entscheidende Rolle zur Reinigbarkeit bei. Beispielsweise können durch eine mangelnde Oberflächenglätte, unsaubere Schweißnähte, ungeeigneten Dichtungskonstruktionen sowie nachträgliche Einbauten massive Probleme bei der Reinigung hervorgerufen werden.
5 Reinigungsverfahren
Neben dem manuellen Reinigungsverfahren besteht auch die Möglichkeit eines automatischen Reinigungsverfahrens (CIP-System). Ein CIP-Reinigungssystem ist allerdings in vielen Fällen nicht realisierbar und zu teuer, weshalb manuell gereinigt werden muss. Dieses ist gegenüber dem CIP-Reinigungssystem hin sichtlich der Reproduzierbarkeit und Validierbarkeit deutlich im Nachteil. Die manuelle Reinigung ist in Ihrer Durchführung stark von der jeweiligen Person abhängig. Deshalb ist nicht nur eine regelmäßige Schulung und die richtige Arbeitsmotivation des Personals, sondern auch die Notwendigkeit einer detaillierten Anweisung, Planung und Überwachung für den Reinigungserfolg entscheidend. Das CIP-System stellt einen kontinuierlichen Prozess dar, der reproduzier- und standardisier bar ist. Der Nachteil liegt darin, dass der Reinigungserfolg aufgrund des geschlossenen Systems schlecht inspizierbar ist. Für die optische Kontrolle des Reinigungserfolgs sowie die Probenahme des Reinigungswassers müssten Bullaugen und Probenahmestellen eingeplant werden. Die Reinigbarkeit von CIP-Systemen kann auch im Rahmen der Qualifizierung mittels einer Bestimmung des Rückstands von Riboflavin erfolgen. Riboflavin weist eine gute Detektierbarkeit auf und ist zudem unbedenklich. Zu bedenken ist auch, dass viele weitere Schritte wie eine Programmierung von Verfahrensparametern, eine Zugriffssicherung oder Archivierung der Daten unternommen werden müssen. CIP-Systeme sind stationäre Anlagen, die fest in die Produktionsanlage integriert sind. Alle auf der Produktionsanlage relevanten Prozesse und Produkte müssen dementsprechend auf das System eingestellt werden.

Die kritischen Mess-, Steuer-, und Regeleinheiten müssen im CIP-System überprüft und regelmäßig rekalibriert werden. Mit Hilfe der in den CIP-Systemen installierten Sprühkugel wird die gesamte Oberfläche der Anlage mit Reinigungslösung benetzt. Die Kugel kann statisch oder drehbar sein. Die Durchflussmenge des Reinigungsmittels ist über den Druck so einzustellen, dass eine Zerstäubung der Lösung vermieden wird. Statisch und dynamisch auftretende Druckverluste können mit einer variabel festgelegten Pumpleistung vermieden werden.
Chemische Eigenschaften des Reinigungs mittels Oft wird mit Wasser gespült, um die Reinigungsmittelanalytik zu umgehen. Das Reinigungsergebnis ist oft schlecht oder sehr viel Wasser wird verbraucht. Hier lohnt sich ein Blick auf die Chemie des zu reinigenden Stoffes. Manchmal kommt eine Komponente (Hilfsstoff) aus der Formulierung in geringer Konzentration als Reinigungszusatz in Frage. Die folgenden Effekte können zum Beispiel für die Reinigung im wässrigen Milieu genutzt werden:
- pH-Verschiebung bei Arzneistoffen, die protonierbar sind
- angehobener Salzgehalt erhöht Löslichkeit schwerlöslicher Stoffe (Aktivität wird verringert)
- Lösungsvermittler/Emulgatoren aus Formulierung von Emulsionen
Der Vorteil dieser Vorgehensweise ist, dass keine zusätzlichen Komponenten zugesetzt werden und damit die Anlage nicht mit zusätzlichen Chemikalien belastet wird, die wieder nachgewiesen werden müssen.
6 Probenahme
In den Plänen zur Reinigungsvalidierung müssen die Orte der Probenahme und eine Begründung für die Auswahl der Orte beschrieben werden. Bei der Probenahme werden zwei an erkannte Arten unterschieden: die direkte und die indirekte Probenahme.
Bei der direkten Probenahme handelt es sich um den Wischtest (Swab), bei dem mittels Wattestäbchen eine definierte Oberfläche mit einem geeigneten Lösungsmittel beprobt wird. Voraussetzung ist hier die Zugänglichkeit der Probenahmestellen worin auch der wesentliche Nachteil liegt: es wird nicht die gesamte Oberfläche beprobt und man kann nicht davon ausgehen, dass die Verunreinigung gleichförmig in einer Anlage verteilt ist. Zudem weist diese Art der Probenahme eine geringe Reproduzierbarkeit auf. Der Vorteil liegt darin, dass man das Ergebnis aus der Analytik direkt einer definierten Stelle in der Anlage zuordnen kann. Auch können schwer lösliche Rückstände mit dem Wischtest erfasst werden.
Die indirekte Probenahme (Rinse) wird bei nicht oder schwer zu erreichenden Stellen, wie Rohrleitungen aber auch Behälterinnenoberflächen und Einbauten, eingesetzt. Der Nach teil der Methode liegt in der Unsicherheit ob alle Rückstände ausgespült worden sind, ob die Rückstände wasserlöslich sind und die Abtragung auch an schlecht zugänglichen Stellen erfolgt. Ein wesentlicher Vorteil liegt je doch in der Möglichkeit eine große Oberfläche zu beproben. Ein weiterer Nachteil der Rinse Methode ist es im Allgemeinen, dass die nach zuweisenden Substanzen stark verdünnt wer den und es dadurch schwer wird, den berechneten Grenzwert analytisch nachzuweisen. Eine weitere Möglichkeit der Probenahme besteht in der Kombination beider Methoden, z.B. eine Beprobung mittels Swab an Stutzen und einer anschließenden Rinse-Beprobung des Gesamtinnenraums. Für beide Probenahmearten ist jeweils die Wiederfindungsrate zu bestimmen.
7 Analytische Methoden
Der Reinigungserfolg einer Methode muss mit analytischen Methoden belegt werden. Im FDA „Guide to inspections validation of cleaning processes“ [5] wird die Notwendigkeit der Spezifität und Sensitivität der analytischen Methoden gefordert. Dabei muss immer beachtet werden, dass Ergebnisse unterhalb der Nachweisgrenze nicht bedeuten, dass in der Probe keine Rückstände vorhanden sind. Das Ergebnis kann nur so gut sein wie die Empfindlichkeit, Spezifität und Genauigkeit des Tests. Daher müssen die analytischen Methoden validiert werden. Folgende Analysenmethoden werden üblicherweise in der Reinigungsvalidierung verwendet.
Spezifische Analysen
- HPLC
- GC
- Farbtests /Fertigkits
- Farbreaktionen auf Proteine (z.B. BCA)
Summenparameter
- TOC
- Leitfähigkeit
- UV/VIS-Spektroskopie
7.1 Analytik für Produkte
HPLC/GC-Analyse: Während der Vorteil der spezifischen Analyse sicherlich in der Spezifität für eine bestimmte Substanz (Wirk- oder Leitsubstanz) liegt, so ist diese Methode in der chemischen Industrie nur für Endprodukte, bzw. nur für den Schritt anwendbar, in dem der Wirkstoff synthetisiert wurde. Daher ist wie bereits der FDA-Guide betont eine spezifische Analyse in der Wirkstoffproduktion nicht praktikabel.
Die spezifischen Methoden haben den Vorteil, dass gezielt spezifische Produkte / Reinigungsmittelkomponenten nachgewiesen werden können. Der Nachteil liegt allerdings darin, dass bei manchen Reinigungsverfahren durch chemische Degradation die tatsächliche vorher vorhandene Menge nicht mehr nachgewiesen werden kann. Hier kann man dann für Analytik der Reinigungsvalidierung eine unspezifischere Bestimmung eines Summenparameters ein setzen.
Falls das Reinigungsverfahren den Wirkstoff zersetzt, wie es z.B. bei der Reinigung von Proteinrückständen mit Natronlauge der Fall ist, reicht eine Risikoanalyse, die den Denaturierungsprozesse bewertet, evtl. kombiniert mit einer Abreicherungstudie.
7.2 Grenzen der Messmethoden
Wenn Säuren oder Laugen als Reinigungsmittel (Zusatz) eingesetzt werden, wird oft der Summenparameter Leitfähigkeit als Meßkriterium für die Abreicherung des Reinigungsmittels verwendet. Daraus ergeben sich folgende Fragen:
- Welche Aussagefähigkeit hat diese Messmethode hier?
- Wenn nach Reinigung mit Purified Water gespült wird und die Leitfähigkeit von Purified Water wieder erreicht ist, sind dann al le Reinigungsmittelrückstände auf ein vertretbares Maß entfernt worden?
Hier muss man im Vorfeld die Grenzen dieses Messverfahrens betrachten und entsprechend auslegen. Das folgende Rechenbeispiel soll die Grenzen des Verfahrens erläutern. Aus den Tabellenwerken [7] ergibt sich eine Nachweisbarkeit von Säuren und Laugen aus den Leitfähigkeiten der Säuren und der maximal zu erwartende Blindwert aus dem Grenzwert für die Leitfähigkeit von Purified Water. Ein Konzentrationsgrenzwert, der kleiner ist, als die Konzentration die sich aus der Annahme ergibt, dass die gesamte Leitfähigkeit am Grenzwert von purified water von dem Reinigungsmittel stammt, gilt als nicht einsetzbar. Als Beispiel soll eine NaOH-Konzentration in WfI berechnet werden. Für Wasser für Injektionszwecke beträgt der Grenzwert der Leitfähigkeit bei 20°C 1,1 µS/cm. Die Leitfähigkeit von NaOH in einer 0,5%igen Lösung beträgt 24,8 µS/cm. Daraus ergibt sich eine Restkonzentration NaOH im WfI von 0,22 mg/l, was ei nem pH-Wert von 8,7 entspricht und damit die Spezifikation von WfI verletzt und daher ungeeignet als Detektionskriterium ist. Das oft verwendete Verfahren „Kessel voll laufen lassen“ würde zu hohe Flüssigkeitsmengen beinhalten und die Aussagekraft der Leitfähigkeitsmessung wäre nicht gegeben. Etwas bessere Nachweisgrenzen erreicht man, wenn man mit Wasser einer geringeren Leitfähigkeit spült und folgende Vorgehensweise einhält. laufen lassen“ würde zu hohe Flüssigkeitsmengen beinhalten und die Aussagekraft der Leitfähigkeitsmessung wäre nicht gegeben. Etwas bessere Nachweisgrenzen erreicht man, wenn man mit Wasser einer geringeren Leitfähigkeit spült und folgende Vorgehensweisen einhält.
- Blindwert Wasser vom aktuellen Spülgang vermessen
- Als Vergleichswert: auf Grenzwert mit Spülwasser verdünnte Reinigungslösung einsetzen.
8 Validierung
Wie in jeder Validierung müssen die Daten aus mehreren Läufen in der Vergangenheit zur Validierung herangezogen werden. Üblicherweise beginnt man mit der Auswertung von drei Läufen und erweitert die Datenlage mit regelmäßigen, geplanten Revalidierungsläufen. Für die Planung der Validierung sind auch Standzeiten einzubeziehen, die sich am geplanten Produktionsablauf orientieren sollten. Seit 2015 [4] ist es zulässig, schon nach dem ersten Validierungslauf mit Verifizierung der Reinigungserfolges die produzierte Charge in den Markt zu bringen. Dies bedeutet insbesondere für Prüfpräparate eine Erleichterung.
9 Fazit
Reinigungsvalidierung ist mehr als „Spülen“, sie erfordert die genaue Betrachtung und Bewertung der einzelnen Schritte und ihrer Leistungsfähigkeit und erfordert ein hohes Maß an interdisziplinärem Wissen
Lorem ipsum dolor sit amet consectetur. Viverra feugiat iaculis aliquam eget sapien sit purus et ultricies. Iaculis nulla egestas risus a. Eget ornare facilisi amet id morbi. Est lectus quisque at vel. In nisi vitae adipiscing augue ante tellus. Enim neque curabitur accumsan tortor senectus. Neque cras fringilla lacinia tristique quis in erat massa.
Literatur
1
ALLHENN, D ; ANHALT, E: Auswahl von Akzeptanzkriterien fr die Reinigungsvalidierung von Mehrzweckanlagen. In: Pharm. Ind. 77 (2015), Nr. 7, S. 1074 1080
2
EUROPEAN COMMISSION: EU Guidelines to Good Manufacturing Practice, Volume 4, Annex 1: Manufacture of Sterile Medicinal Products. 2008
3
EUROPEAN MEDICINES AGENCY: Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities. 2014
4
EUROPEAN COMMISSION: EU Guidelines to Good Manufacturing Practice, Volume 4, Annex 15: Qualification and Validation. 2015
5
FOOD AND DRUG ADMINISTRATION: Guide to Inspections- Validation of Cleaning Processes, 1993
6
FOURMAN, G L. ; MULLEN, M: Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations. In: Pharmaceutical Technology (1993), Nr. 4, S. 54–60
7
HAYNES, William M. (Hrsg.): CRC Handbook of Chemistry and Physics. 91. Baca Raton, FL, 2010
8
HEBENBROCK,K ;HRACH, J: Interner Bericht DPhE: Toxikologisches Assessment von Bupivacain- Ermittlung der PDE. 2015
9
ICH: Technical Requirements for Registration of Pharmaceuticals for Human Use: Q9- Quality Risk Managment. 2004
10
ICH: Technical Requirements for Registration of Pharmaceuticals for Human Use: Q3C Impurities: Guideline for Resi dual Solvents. 2018
11
PIC/S: Validation Masterplan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation. 2007
12
US FOOD AND DRUG ADMINISTRAI ON: Code of Federal Regulations Title 21, PART 184 DIRECT FOOD SUBSTANCES AFFIRMED AS GENE RALLY RECOGNIZED AS SAFE.– URL https://www.accessdata.fda.gov/ scripts/cdrh/cfdocs/cfcfr/ CFRSearch.cfm?CFRPart=184